
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrgan preservation with targeted rapamycin nanoparticles: a pre-treatment strategy preventing chronic rejection in vivo
Peng Zhuabc,
Carl Atkinson†
ab,
Suraj Dixitd,
Qi Chengabc,
Danh Tranb,
Kunal Patelab,
Yu-Lin Jiangd,
Scott Esckilsenab,
Kayla Millerd,
Grace Bazzleb,
Patterson Allenab,
Alfred Moore‡
d,
Ann-Marie Broome†‡ *de and
Satish N. Nadig†*ab
*de and
Satish N. Nadig†*ab
aDepartment of Surgery, Division of Transplant Surgery, Medical University of South Carolina, USA. E-mail: nadigsn@musc.edu
bDepartment of Microbiology and Immunology, Lee Patterson Allen Transplant Immunobiology Laboratory, Medical University of South Carolina, USA
cInstitute of Organ Transplantation, Department of Surgery, Hepatic Surgery Center, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
dDepartment of Cell and Molecular Pharmacology & Experimental Therapeutics, Medical University of South Carolina, USA. E-mail: broomea@musc.edu
eDepartment of Bioengineering, Clemson University, USA
First published on 23rd July 2018
Abstract
Hypothermic preservation is the standard of care for storing organs prior to transplantation. Endothelial and epithelial injury associated with hypothermic storage causes downstream graft injury and, as such, the choice of an ideal donor organ preservation solution remains controversial. Cold storage solutions, by design, minimize cellular necrosis and optimize cellular osmotic potential, but do little to assuage immunological cell activation or immune cell priming post transplantation. Thus, here we explore the efficacy of our previously described novel Targeted Rapamycin Micelles (TRaM) as an additive to standard-of-care University of Wisconsin preservation solution as a means to alter the immunological microenvironment post transplantation using in vivo models of tracheal and aortic allograft transplantation. In all models of transplantation, grafts pre-treated with 100 ng mL−1 of TRaM augmented preservation solution ex vivo showed a significant inhibition of chronic rejection post-transplantation, as compared to UW augmented with free rapamycin at a ten-fold higher dose. Here, for the first time, we present a novel method of organ pretreatment using a nanotherapeutic-based cellular targeted delivery system that enables donor administration of rapamycin, at a ten-fold decreased dose during cold storage. Clinically, these pretreatment strategies may positively impact post-transplant outcomes and can be readily translated to clinical scenarios.
Introduction
Improvements in immunosuppressive medications have allowed for better short-term outcomes in all aspects of solid organ transplantation. However, over the past two decades there have been few advances in the prevention of chronic transplant dysfunction (CTD). CTD remains a leading cause of graft loss in the long term and many of its effects can be traced back to the initial oxidative and immunological insults accrued during early ischemia reperfusion injury to endothelial and epithelial cells post-reperfusion.Central to these immunological insults is the activation of donor cells that, upon brain death, organ procurement, organ preservation, and reperfusion, promotes inflammation, cytokine and chemokine release and is central to the programming of recipient immune cells. It is appreciated that modulation of the endothelium prior to transplantation may improve graft outcomes.1,2 Recent studies have shown that treatment of endothelial cells (ECs) in vitro with the mTOR inhibitor rapamycin, an immunosuppressive drug used clinically, can render ECs tolerogenic.3 Pre-treatment of ECs with rapamycin reduced proliferation of allo-reactive memory T cells, cytokine production, and EC activation. Pre-treatment further promoted the differentiation of T regulatory cells in an EC/T cell co-culture system.1,3–5 These elegant studies demonstrate that pre-injury rapamycin therapy provides protection from EC-mediated immune injury. Furthermore, in in vivo studies, human aortic grafts were transplanted into immunodeficient SCID/beige mice and grafts were allowed to “heal-in” for 30 days, after which, mice were treated with rapamycin for 3 days.3 Grafts were then harvested and re-transplanted into humanized SCID/beige mice. Grafts from rapamycin pre-treated donors were significantly protected from graft injury and chronic rejection, as compared to untreated controls.3 The conclusion from these studies was that rapamycin-modulated donor ECs allowed for protection against the development of CTD by reprogramming their ability to present antigen and blunt initial responses to injury post-reperfusion. Although rapamycin has been touted to confer a protective and tolerogenic phenotype to transplant recipients, its use in the perioperative period is hindered by the drug's numerous systemic side effects.6 However, given its clinical promise, strategies that provide a platform to facilitate donor rapamycin delivery in a relevant time frame are necessary if these results are to be translated clinically.7,8
Here, we capitalized on these unique findings and have developed a bioengineered solution delivering rapamycin to the donor organ prior to transplantation, by the addition of Targeted Rapamycin Micelles (TRaM) to standard organ preservation solution. Micellar nanoparticles (NPs) are ideal vehicles for therapeutics due to their small size and hydrophobic core allowing for self-assembly with hydrophobic payloads such as rapamycin. Additionally, micellar constructs facilitate the application of surface targeting moieties that can be tailored to promote uptake by specific cell types or under specific physiochemical conditions. To this end, we have previously designed, constructed, and characterized rapamycin micelles decorated with cyclic arginine–glycine–aspartate moieties (cRGD) specific for alpha V beta 3 (αVβ3) integrins located on the surface of endothelial and epithelial cells. Although αVβ3 integrin molecules are not specific for endothelial or epithelial cells, using these TRaMs, we demonstrated that membrane targeting facilitated receptor-mediated endocytosis and significantly enhanced micelle-specific uptake as compared to untargeted constructs in in vitro models.9 Further, we have shown that TRaMs inhibit EC cytokine production and alloreactive T cell proliferation, as compared to free rapamycin or untargeted rapamycin micelles, in in vitro models.9
Most transplanted organs in the United States are procured from deceased donors, and as such, the organ must be cold stored for a significant period of time until it can be transplanted into a suitable recipient. Various preservation solutions exist, each substantially different in their composition, but the purpose of each are similar: to prevent cellular edema, delay cell destruction, maintain organ metabolic potential, and maximize organ function after perfusion is re-established. They do nothing to modulate the immunologic injury suffered as a consequence of organ donation nor do they prepare the donor organ for the oncoming immunological attack by the recipient's immune system. This necessary step in the transplantation process provides a window of opportunity to deliver therapeutics directly to the vascular endothelia, and/or the respiratory epithelia of the donor organs ex vivo as an additive to a standard preservative such as University of Wisconsin (UW) solution.10,11
Therefore, in this study, we report the application of our novel TRaM nanoparticles as an additive to UW and explore the impact of donor organ nanoparticle delivered rapamycin on the development of chronic rejection.9 For the first time, we show that donor organ pre-treatment with TRaM augmented UW preservation solution, as compared to standard-of-care UW alone and UW augmented with free rapamycin, protects against the development of chronic rejection in two separate in vivo allograft models.
Methodology
Preparation of micelles
Micelle encapsulation of rapamycin (RaM) was carried out as described by Nadig et al.9 Rapamycin was mixed with amino-PEG-PE 1,2-diacyl-sn-glycero-3-phosphoethanolamine-N-[amino-poly(ethylene glycol)] and PHC (N-palmitoyl homocysteine (ammonium salt)), suspended in chloroform. The chloroform mixture was sonicated then the solvent was evaporated in a vacuum oven at room temperature. The pellet obtained after evaporation was heated to 80 °C and dissolved in nanopure water to produce amine-functionalized micelles. The micelle solution was sonicated in a water bath and filtered to remove aggregates. For the synthesis of TRaM, the RaM solution was used for peptide conjugation (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of carboxyl group on peptide to amine group on the micelles at 30% coverage of amines). cRGD was added to MES buffer (pH 4.5) in separate scintillation vials followed by EDC and sulfo-NHS. PBS (pH ∼12) was added to bring the pH back to 7.5. The micelle solution was added to the peptide solution and incubated at room temperature. Excess peptide was removed using 10 K MWCO ultracentrifugal device (Millipore, MD). For dye labelling, NHS Dylight 680 was added at a ratio covering 30% amine groups of the micelles to RaM and TRaM, respectively. Excess dye was removed using 10 K MWCO ultracentrifugal device.
:1 ratio of carboxyl group on peptide to amine group on the micelles at 30% coverage of amines). cRGD was added to MES buffer (pH 4.5) in separate scintillation vials followed by EDC and sulfo-NHS. PBS (pH ∼12) was added to bring the pH back to 7.5. The micelle solution was added to the peptide solution and incubated at room temperature. Excess peptide was removed using 10 K MWCO ultracentrifugal device (Millipore, MD). For dye labelling, NHS Dylight 680 was added at a ratio covering 30% amine groups of the micelles to RaM and TRaM, respectively. Excess dye was removed using 10 K MWCO ultracentrifugal device.
In order to assess the quality of micelles and obtain quantitative measures that allow comparison between different batches of micelles, various parameters are monitored. For micelles used in analytical and bioanalytical applications, the main characteristics include the average diameter and degree of size polydispersity; encapsulation efficiency; the ratio of phospholipids to encapsulant concentration; lamellarity determination. Small batch-to-batch variations in the amine-PEG-PE and PHC ratio resulted in small differences in their physical parameters. For experimental purposes, only batches with >95% similarity were used.
HPLC analysis of rapamycin encapsulation in micelles
Analysis of standard rapamycin and rapamycin in micelle samples were performed with a Dionex UltiMate3000 chromatograph (Thermo Scientific) equipped with a UV-visible detector and an auto sampler as described with minor modifications.12,13 Rapamycin was separated on a C18 reverse phase analytical column (250 mm × 4.6 mm 5 micron particles) under isocratic condition. The mobile phase was a mixture of acetonitrile and water in the ratio 98:2 (v/v). The flow rate was 1 mL per minutes and rapamycin was monitored at 278 nm with retention time of 4.8 minutes at 21 °C.
Hypoxic cold storage models of ischemia-reperfusion injury
Mouse cardiac endothelial cells (MCEC) (Cedarlane, Ontario, Canada), were cultured to confluency in Dulbecco's Modified Eagle Medium (DMEM) (Life Technologies, CA) in a 37 °C incubator with humidified room air and 5% CO2. Upon achieving confluency MCEC were exposed to simulated cold storage, and reperfusion injury, as previously described.9 In brief, media was replaced with ice cold University of Wisconsin solution (Bridge to Life, SC), augmented with either rapamycin, cyclosporine (CSA), FK506 (tacrolimus; TAC), or mycophenolate mofetil (MMF) at three different doses; 1, 10 and 100 ng mL−1. Cells were rendered hypoxic in a hypoxic chamber (Billups-Rothenberg Inc, CA), flushed with pure nitrogen (Airgas, PA), and subjected to hypoxic condition for 6 hours at 4 °C. After cold hypoxic ischemia, cells underwent 24 hours of simulated reperfusion by removing the preservation solution and reintroducing warm culture media, and under normal culture conditions. To assess the impact of immunosuppressive regimes on pro-inflammatory cytokine release and cell injury, supernatants were removed to assay for KC release by standard ELISA techniques (BD Biosciences, NJ) and MTS Assay (Sigma Aldrich, MO), respectively.In vitro analysis of mTOR inhibition
To assess the efficacy of TRaM and free rapamycin on inhibiting MCEC mTOR activity, MCECs were again exposed to simulated cold hypoxic reperfusion injury, using the model outlined above. In these experiments, UW solution was augmented with TRaM, empty micelles, free rapamycin, or DMSO vehicle. Following 6, 12, 18, 24, or 72 hours of reperfusion, MCEC protein lysates were collected and analyzed by western blot as previously described.14 Briefly, cells were washed, detached mechanically, pelleted and lysed with M-PER Mammalian Protein Extraction Reagent (Thermo Fisher Scientific, MA) that contained 1× phosphatase and 1× protease cocktail inhibitors (Thermo Fisher Scientific, MA). After quantification, proteins were denatured and separated using 4–20% gradient SDS-PAGE (Bio-Rad Laboratories, CA), followed by wet transfer to a PVDF membrane (Bio-Rad Laboratories, CA) for immunoblotting. Primary antibodies against P-p70S6K (Thr389), p70S6K (all from Cell Signaling, MA), beta actin (Santa Cruz, TX) were used at a 1:1000 dilution. Secondary antibodies conjugated to horseradish peroxidase (GE Healthcare, Little Chalfont, UK) were used at a 1:2000 dilution. Signal was determined by chemiluminescence using film development and digital imaging methodologies. Quantification of band intensity was performed using ImageJ v1.46 (NIH, MD).
In vitro and ex vivo uptake of nanoparticles
We have previously demonstrated the uptake of TRaMs in MCECs and aortas, and thus here the in vitro and ex vivo uptake of TRaMs was assessed in human bronchial epithelial cells (BEAS-2B) and mouse trachea.11 For in vitro experiments BEAS-2B cells were exposed to simulated cold hyperoxic storage and reperfusion as previously described.15 Cells were exposed to UW or UW augmented with TRaM and following 18 hours of cold storage, were reperfused for 10 and 60 minutes to visualize TRaM internalization using Olympus Fluoview FV10i LIV Confocal Microscope (Olympus, NC). Donor tracheas were harvested for analysis of ex vivo delivery of NPs. Tissue segments were stored on ice for 6 hours in UW solution, or UW solution augmented with increasing doses of RaM and TRaM (100, 500, and 1000 ng mL−1). Fluorescence multispectral imaging was performed using the Maestro Imaging System (PerkinElmer, MA). Multispectral images were acquired under a constant exposure of 2000 ms with an orange filter acquisition setting of 630–850 nm in 2 nm increments. Multispectral images were unmixed into their component spectra (Dylight 680, autofluorescence, and background) and these component images were used to gain quantitative information in terms of average fluorescence intensity by creating regions of interest (ROIs) around the organs in the Dylight 680 component images.Animals
All laboratory animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of The Medical University of South Carolina (MUSC). Experiments were approved by the by the Division of Laboratory Animal Resources (DLAR) under the direction of Suzanne Craig, D. V. M., DACLAM, MBA. An assurance statement is on file with OPRR/DHHS detailing program for laboratory animal care at MUSC. MUSC has ongoing full accreditation from AAALAC effective November 5, 1987. MUSC has an approved assurance from OLAW-NIH (A3428-01) until April 30, 2020.Male Balb/c and C57BL/6 mice (8–12 weeks) were obtained from Jackson Laboratory (Bar Habor, ME). All mice used for transplantation studies weighed 20–30 g and were housed under specific pathogen free conditions at MUSC. For all surgical procedures, mice were anesthetized with intraperitoneal injections of ketamine (75 mg kg−1; Lloyd Laboratories, IA) and xylazine (16 mg kg−1; Vedco, Inc., MO).
Tracheal transplantation
Orthotopic tracheal transplants were performed as previously described.16,17 Balb/c donor trachea, 6–8 tracheal ring segments, were excising and stored in either control UW solution or treatment groups consisting of TRaM, RaM, free rapamycin, or empty micelles at 1000 ng mL−1. All donor tracheas were stored in their respective solutions for 4 hours on ice prior to transplantation into allogeneic C57Bl/6 recipients. Tracheal grafts were excised on days 7 and 28 days post transplantation for analysis.Aorta transplantation
Balb/c donor mice were anesthetized and infrarenal aorta isolated and cuffs secured at both ends with 10-0 nylon suture (AROSurgical Instruments Corporation, Newport Beach, CA). The donor aorta were then stored on ice in University of Wisconsin (UW) solution alone (Bridge to Life Ltd, SC), or UW augmented with either TRaM, RaM, free rapamycin, or empty micelles at dose of either 100 or 1000 ng mL−1. All donor aortas were stored in their respective solutions for 6 hours prior to transplantation in C57Bl/6 allogeneic recipients.Transplantations were performed as previously described using a modified-cuff technique18 and grafts harvested 28 days post transplantation.
Histopathology
Allografts were harvested at post-operative days 7 or 28 and tissue segments were embedded in paraffin. Six μm thick aorta or trachea sections were stained with hematoxylin and eosin and Elastin-van Gieson stain (EvG, HT25; Sigma, MO). Morphometric analysis of transplant arteriosclerosis on EvG stained sections was measured as described.19 Tracheal graft injury and remodeling was determined as previously described.20Statistical analysis
All data are expressed as mean +/− SD. All data analysis was performed using GraphPad Prism software version 6 (La Jolla, CA) unless specified. Multiple variables were analyzed via analysis of variance techniques, and a p value < 0.05 was considered statistically significant.Results
Rapamycin exhibits improved immunosuppressive capacity when compared to current standard-of-care regimens
In order to establish the superiority of mTOR inhibition over standard-of-care immunosuppressants, we performed a head-to-head comparison of rapamycin to cyclosporine (CSA), FK506 (tacrolimus; TAC), and mycophenolate mofetil (MMF). Using mouse keratinocyte chemoattractant (KC), a surrogate for IL-8 in the murine system and a potent inflammatory cytokine of ECs and epithelial cells, as a readout. ELISAs were performed in a dose-escalating manner with no drug and vehicle as controls. Rapamycin reduced levels of EC released mouse KC across three varying doses and significantly suppressed KC levels when compared to MMF and TAC at 1, 10 and 100 ng mL−1, respectively (Fig. 1a). All immunosuppressives were tested for their toxicity profile against mouse ECs and no significant toxicity was noted across each of these doses (Fig. 1b).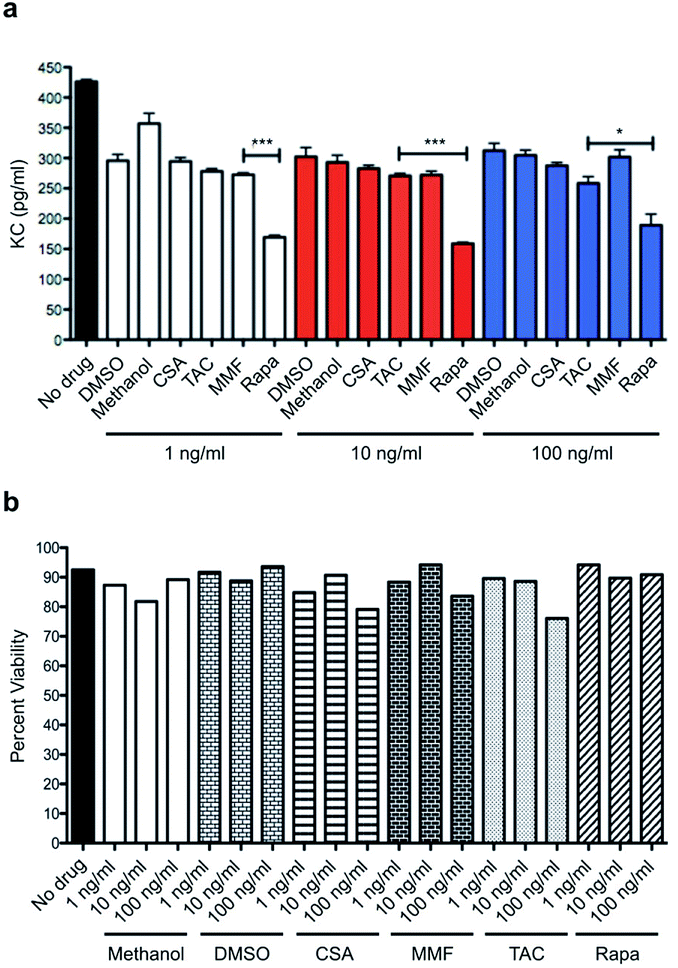 | ||
| Fig. 1 Comparison of SOC immunosuppressants to rapamycin. (a) In mouse endothelial cells treated with H2O2, mouse KC, an analogue of IL-8, is significantly suppressed by rapamycin when compared to conventional immunosuppressive modalities such as cyclosporine (CSA), tacrolimus (TAC), or mycophenolate mofetil (MMF). This effect is sustained amongst varying and escalating doses. (b) MCEC treated with increasing concentrations of each immunosuppressive did not impact cell viability. *p < 0.05; ***p < 0.05. | ||
Preconditioning MCECs with rapamycin inhibits mTORC1 activity
We utilized a novel in vitro system of ischemia reperfusion wherein mouse cardiac ECs (MCEC) were subjected to cold ischemia (hypoxic chamber) for 6 hours in UW solution alone or UW solution augmented with 100 ng mL−1 free rapamycin. Following the ischemic insult, the cells were reperfused with warm media to mimic reperfusion. EC lysates were obtained at specific post-reperfusion time points and ribosomal protein S6 kinase beta-1 or P-p70S6K (Thr389) detected by western blotting to determine mTOR activation as the phosphorylation of this ribosomal protein at threonine 389 is a well-described marker for activation by mTOR.In rapamycin treated cells, P-p70S6K is completely absent at 6, 12, 18, and 24 hours post-reperfusion indicating pre-treatment maintains its inhibitory effects 24 hours post-reperfusion (Fig. 2a and b). When rapamycin was added after cold storage upon reperfusion in warm media for 6, 12, 18, 24 hours P-p70S6K remained absent (Fig. 2c and d).
 | ||
| Fig. 2 Preconditioning of EC with rapamycin compared to treatment with rapamycin post-reperfusion. MCECs were subjected to hypoxic cold storage for 6 hours in UW solution alone or UW solution augmented with 100 ng mL−1 rapamycin, followed by warm reperfusion. Whole cell lysates were obtained and immunoblotted for P-p70S6K (Thr389). (a) A representative immunoblot showing P-p70S6K is completely absent during the first 24 hours post-reperfusion. (b) Band intensity was normalized to beta-actin. Another group of MCECs was subjected to hypoxic cold storage for 6 hours in standard UW solution, followed by warm reperfusion with media augmented with 100 ng mL−1 rapamycin for 6, 12 or 24 hours. Whole cell lysates were obtained and immunoblotted as above. (c) P-p70S6K is completely absent at 6, 12, and 24 hours post-reperfusion in both scenarios of rapamycin treatment during or after cold storage. (d) Band intensity of the representative immunoblot normalized to beta-actin. | ||
Bioengineering rapamycin micelles
Micelles encapsulating rapamycin, both untargeted (RaM) and targeted (TRaM), were prepared as previously described.11 TRaM are relatively monodisperse and measure 15.3 nm +/− 2.3 nm (PDI 0.03). Encapsulation efficiency (EE) is approximately 42% and the zeta potential is measured as −40.64 +/− 3.93 mV. The micelles are relatively stable under various physiological conditions, including buffered solutions and serum containing solutions. Over time, only 20–26% of the encapsulated rapamycin leaches from the micelles.Reverse phase-high liquid chromatography (RP-HPLC) was used to analyze solubilized rapamycin in the phospholipid-based vehicle.12,13 Separation was performed on a C18 column reverse phase analytical column under isocratic conditions with a mixture of acetonitrile and water as the mobile phase. The separation process resulted in reasonable retention times and successful separation of the analytes (Fig. 3a, c and d).
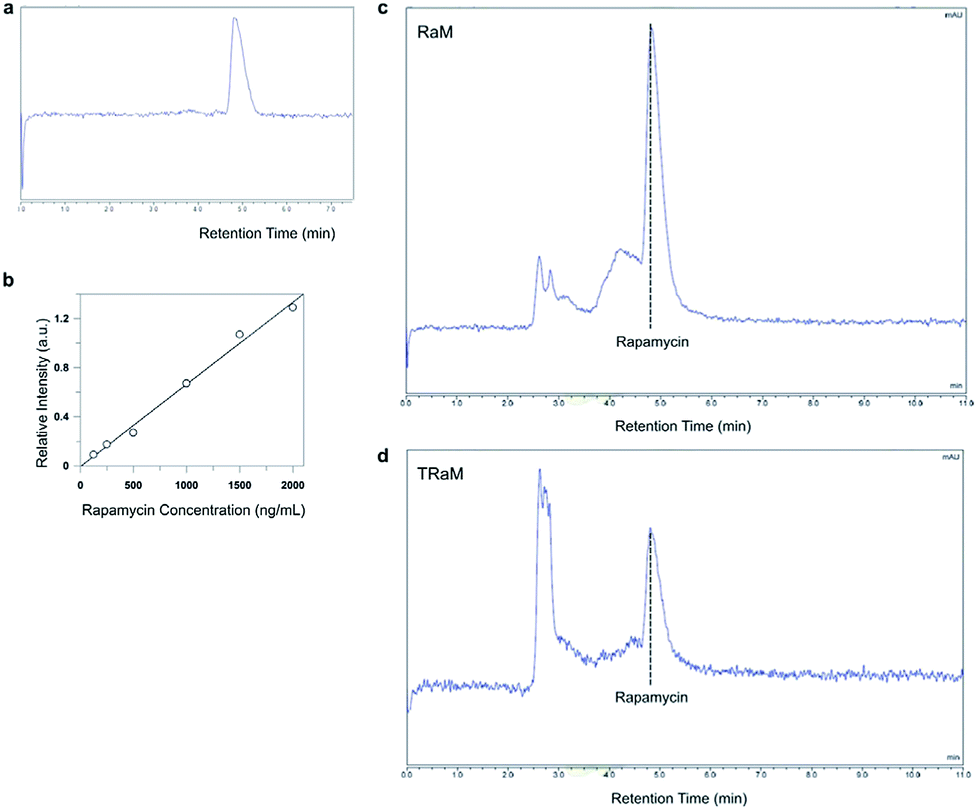 | ||
| Fig. 3 Analysis of rapamycin-encapsulated micelles using RP-HPLC. (a) Separation of purified rapamycin on a C18 column occurred at an approximately 5 minute retention time. (b) Standard curve of rapamycin for the analytical range 125–2000 ng mL−1 (c and d). Representative chromatograms of the successful separation of rapamycin from RaM and TRaM, respectively. | ||
Encapsulation efficiency of rapamycin was calculated at approximately 42% within the micelles. Linearity of the method was confirmed by preparing rapamycin standard curves for the analytical range of 125–2000 ng mL−1 (Fig. 3b). Statistical analysis using least square regression (r2 ≥ 0.99) confirmed linearity for rapamycin for all standard curves.
Functional doses of rapamycin and TRaM nanotherapy inhibit mTORC1 activity equally in MCEC
Utilizing the same in vitro model of ischemia reperfusion, we compared MCEC subjected to cold ischemia for 6 hours in standard UW solution or UW solution augmented with free rapamycin at 1000 ng mL−1, TRaM nanotherapy at 1/10 of the dose of free rapamycin (100 ng mL−1), and empty micelles with no payloads. MCEC were then reperfused with warm media in standard fashion. Whole cell lysates were obtained at 24 and 72 hours post-reperfusion and immunoblotting was performed to detect P-p70S6K (Thr389). TRaM nanotherapy significantly inhibited the activation of mTOR complexes signified by an absence of phosphorylated P70S6K (Fig. 4). | ||
| Fig. 4 Functional inhibition of TRaM (100 ng mL−1) and rapamycin (1000 ng mL−1) were assessed. MCECs were subjected to hypoxic cold storage for 6 hours in standard UW solution or UW solution augmented with TRaM, free rapamycin, empty micelles, or DMSO vehicle, followed by warm reperfusion with culture media. Whole cell lysates were obtained at 24 or 72 hours post-reperfusion, and immunoblotting was performed to detect P-p70S6K (Thr389). (a) A representative immunoblot showing P-p70S6K is completely absent at 24 and 72 hours post-reperfusion. (b) Band intensity of the representative immunoblot normalized to beta-actin. | ||
TRaM-enhanced UW solution allows for nanoparticle uptake in normoxic and cold hyperoxic environments and dampens the inflammatory cytokine response
We have previously shown that TRaM are taken up under cold hypoxic conditions by receptor-mediated endocytosis9 in MCECs, and therefore in order to verify the clinical relevance of the TRaM nanotherapy in lung transplantation, human bronchial epithelial cells were used (BEAS-2B) to interrogate TRaM uptake in cold hyperoxic environments (i.e. cold preservation in inflated lungs) as compared to normothermic, normoxic environments.Here, we subjected human BEAS-2b cells to 4 °C and 100% oxygen environments for 6 hours and then reperfused the cells with warm UW with or without TRaM. Pre-reperfusion the TRaM is isolated to the membrane and proceeds to enter the cells over a period of 60 minutes post-reperfusion (Fig. 5a). BEAS-2B cells were subjected to cold hyperoxia, for 18 hours in standard UW solution or UW solution augmented with the following treatment groups: TRaM, free rapamycin, empty micelles, vehicle, and normal BEAS-2b cells. Following cold storage, the cells were reperfused with warm media and cell lysates obtained at 24 hours post-reperfusion. In keeping with our findings with MCECs (Fig. 4), P-p70S6K (Thr389) expression was attenuated by TRaM cold storage preconditioning 24 hours post-reperfusion (Fig. 5b and c). Furthermore, TRaM pre-treatment was associated with a dampening in epithelial cell inflammation as measured by IL-8 secretion, a key modulator of immune cell migration and activation post lung transplantation (Fig. 5d).
 | ||
| Fig. 5 Accumulation and functional activity of TRaM in vitro. (a) Confocal microscopy reveals TRaM (blue) uptake into epithelial cells over 60 minutes in cold hyperoxia as a surrogate for lung transplantation when compared to normoxic cells (b and c). BEAS-2B cells were subjected to hyperoxic cold storage for 18 hours in standard UW solution or UW solution augmented with 100 ng mL−1 TRaM, 100 ng mL−1 free rapamycin, 100 ng mL−1 empty micelles, or DMSO vehicle, following which the cells were reperfused with warm media. Whole cell lysates were obtained at specific post-reperfusion time points, and P-p70S6K (Thr389) detected by western blot. (b) A representative immunoblot indicating P-p70S6K is completely absent at 24 hours post-reperfusion and (c) band intensity of the representative immunoblot normalized to beta-actin, (d) significant decrease in IL-8 production in rapamycin or TRaM treated BEAS-2b cells compared to those stored in UW alone. | ||
TRaM uptake in tracheal tissues
To directly visualize the ex vivo uptake of TRaM, we administered increasing concentrations of Alexa fluor-680 labelled micelles (TRaM, RaM, and empty) in UW solution to murine tracheas and cold stored them for 6 hours. Following rinsing with UW solution, the tracheas were imaged (Fig. 6a). The ex vivo fluorescent signals increased in a dose and time dependent manner for both RaM and TRaM (Fig. 6b), with cRGD targeted TRaM resulting in greater micelle uptake as compared to RaM or empty micelles with a 3-fold higher fluorescence noted. Overall, these results demonstrate that micelles can successfully accumulate within the trachea during ex vivo cold storage.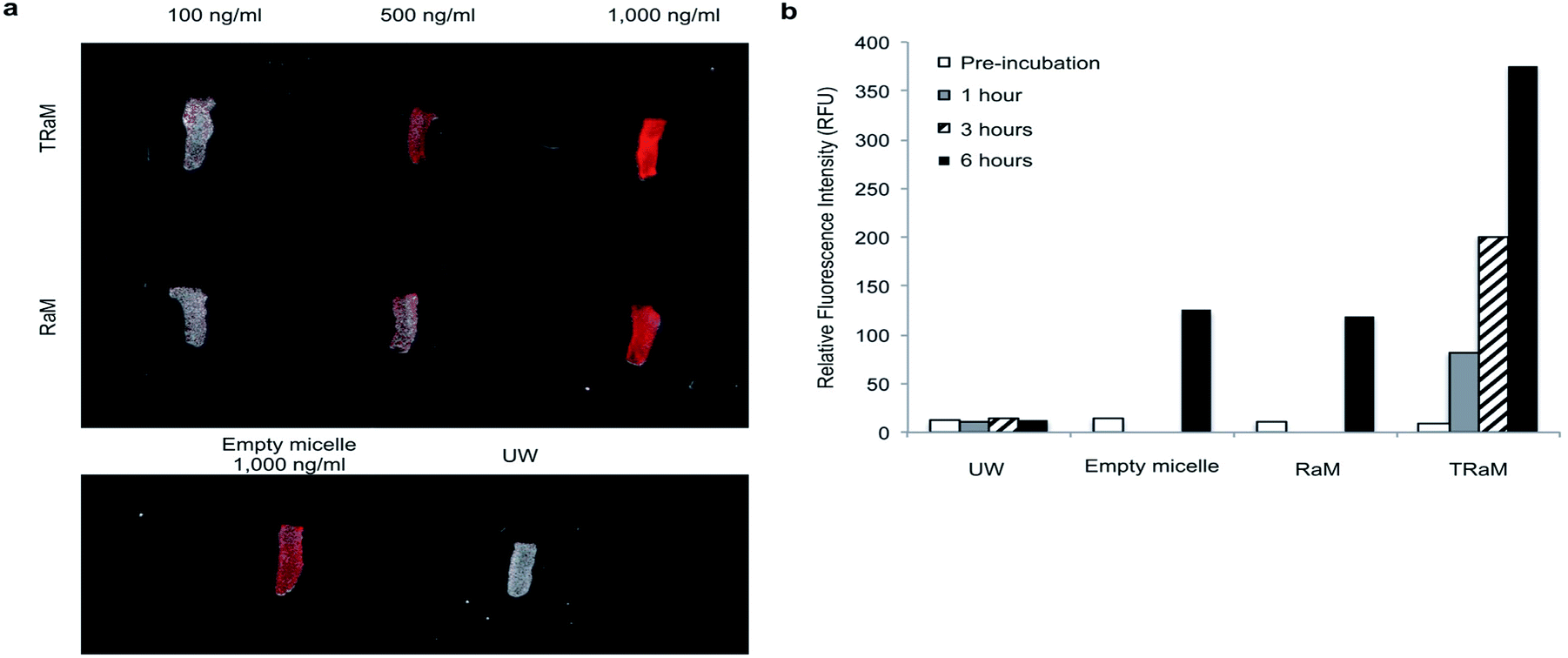 | ||
| Fig. 6 Accumulation and functional activity of TRaM in vivo (a and b). Dose escalation of TRaM and RaM shows increased uptake of nanoparticles in tracheal segments over a 6 hours period as quantified by relative fluorescence intensity. TRaM shows increased uptake when compared to untargeted nanoparticles, empty micelles, or UW alone. | ||
Pre-treatment with TRaM-enhanced UW solution prevents fibrosis in orthotopic tracheal allografts
Utilizing the orthotopic tracheal transplant model as an established microsurgical surrogate for lung transplantation, tracheas were procured from Balb/c mice, and stored in UW solution alone, or UW solution augmented with 1000 ng mL−1 of rapamycin, or 100 ng mL−1 TRaM for 4 hours prior to orthotopic transplantation into fully allogeneic C57/BL6 recipients. Grafts were procured at 7 days (Fig. 7a) and 28 days (Fig. 7b). At both time points, the epithelial to lamina propria ratio and pathological fibrotic score revealed that TRaM-treated tracheas had minimal evidence of disease pathology and were similar to isografts when compared to those tracheas stored in UW solution alone or augmented with free rapamycin (Fig. 7c and d). Of note, recipients of donor trachea cold stored in 1000 ng mL−1 of free rapamycin had an increase incidence of fatal tracheal dehiscence in 6 of 12 transplanted mice. No dehiscence was observed in UW alone or UW solution augmented with TRaM. | ||
| Fig. 7 TRaM pre-treated tracheal allografts after transplantation. (a) Tracheal allografts stored in UW solution alone or UW augmented with 1000 ng mL−1 of free rapamycin, or 100 ng mL−1 of TRaM were transplanted and procured at 7 days (a) or 28 days (b) after implantation. Epithelial/lamina propria ratios (c) and pathologic scores (d) of fibrosis revealed significant protection from fibrotic scarring in trachea treated with TRaM when compared to UW alone or 10-fold more free drug. **p < 0.05; ##p < 0.03. | ||
Pre-treatment with TRaM enhanced UW solution prevents allograft vasculopathy in aortic interposition allografts
The hallmark pathology of chronic graft rejection is represented by the fibro-proliferative lesions that occur in the vessels supplying and within organ allografts. We performed aortic interposition grafts between Balb/c donors to completely allogeneic C57/BL6 mice. Mouse aortas were cold stored in UW solution alone or augmented with 100 or 1000 ng mL−1 of free rapamycin or 10 or 100 ng mL−1 of TRaM for 6 hours. Aortas were transplanted between syngeneic C57/BL6 mice as isograft controls. Twenty-eight days after transplantation, the aortas were procured and H&E and Elastin van Gieson (EvG) staining performed to determine the degree of vasculopathy using previously established formulas.21Animals in the UW solution alone and free drug groups exhibited marked amounts of vasculopathy (Fig. 8a and b). In contrast, a single dose of TRaM at 1/10 (100 ng mL−1) the dose of highest free drug added to UW solution significantly abrogated vasculopathy, with intimal thickening not dissimilar to that seen in isograft controls. However, to confirm its therapeutic nature when doses of TRaM were reduced a further 10-fold, the therapeutic effect was lost supporting the dose-dependent nature of the nanotherapy.
 | ||
| Fig. 8 TRaM pre-treated aortic interposition allografts. (a) EVG staining revealed an absence of vasculopathy in isografts. Animals receiving allotransplants developed marked vasculopathy when the grafts were stored in UW solution alone or pre-treated with 1000 ng mL−1 of free rapamycin. When treated with 10-fold less drug in TRaM and delivered ex vivo as part of the preservation solution, animals exhibited a near complete, significant abrogation in vasculopathy similar to that of control isografts. (b) Doses of free drug and TRaM were decreased 10-fold more breaking the therapeutic effect validating the dose dependency of the nanotherapy. N ≥ 5 animals per group. *p < 0.05. | ||
Discussion
Solid organ allograft survival in the short-term has improved, however success in achieving long-term survival remains elusive. There is growing appreciation that factors occurring early in the life of the graft may affect late outcomes. Expanding evidence suggests that blunting the immunologic activation of donor antigen presenting cells, such as endothelial and epithelial cells, prior to or after transplantation may be the key to downregulating early inflammatory processes which leads to downstream graft loss.22–24 To that end, we have devised a novel nanoparticle that, via a targeting moiety, facilitates nanoparticle uptake in endothelial and epithelial cells in vitro and in vivo. Using these unique characteristics we are able to deliver therapeutic payloads ex vivo and “pre-treat” organs prior to the onslaught of ischemia/reperfusion injury. The prevention of chronic rejection by rapamycin at 1/10 the dose of free drug when encapsulated into a micellar nanoparticle further allows for impactful clinical translation given the host of systemic clinical adverse effects caused by the drug in its current form. In these novel studies, we investigated the efficacy of targeted rapamycin nanotherapy in vitro and then applied it to an in vivo system mimicking bronchial fibrosis in lung transplantation and transplant arteriosclerosis, the hallmark vascular lesion of chronic transplant dysfunction in all solid organ transplants.Various studies suggest that systemic delivery of nanoparticles and emulsions containing chemotherapeutics concentrated in a transplanted heart can prevent vasculopathy.25,26 Yet others advocate for the use of nanoparticles decorated with immunotherapeutics as a delivery device for tissues such as allo-islets to allow for implantation under the shield of immunomodulating therapeutics.27 Pharmacotheapeutic delivery of immunosuppressants via micro and nanoparticles have been touted to represent the “next era” of transplantation, as increased doses of drug may be delivered directly to the area of need. Yet, to date, studies investigating the use of NPs in transplantation have largely revolved around systemic delivery with increased concentration in the grafted tissues and not on treatment of the donor organ themselves before transplant. Although manipulation of donor organs prior to transplantation is not currently the standard-of-care and represents a paradigm shift in transplantation, pre-treatment strategies may be the key to unlocking the early problems that lead to late graft loss. Along with pre-treatment strategies, delivery mechanisms intended to allow for organ level immunosuppression while leaving the patient's global immune system intact may represent the future of organ specific immunosuppression. These disruptive technologies include micro and nanoparticles carrying immunosuppressants and chemotherapeutics, for example, delivered both systemically and directly to an organ allograft.25,26,28
In the studies presented here, we utilized rapamycin as it has been previously shown that rapamycin not only prevents the development of vasculopathy, but is implicated in the induction of tolerance, and maintains a hydrophobic chemical composition allowing it to serve as the core of the micelle. In addition, despite the immunologic advantages of the drug, rapamycin is seldom used in the perioperative period due to its deleterious systemic side effect profile. Investigations wherein human arterial segments transplanted as aortic allografts in rapamycin-treated mice inoculated with allogeneic PBMC are resected and re-transplanted into other humanized mice, revealed an abrogation of T-cell mediated vasculopathy, further attesting to the drug's unique immunologic advantage.3 Although elegant, these studies are not readily translated to the clinic as the “donor” would require rapamycin pre-treatment. However, the effects of rapamycin on decreasing the interferon gamma axis and inhibiting the proliferation of vascular smooth muscle cells and recruitment of effector T cells by up-regulating inhibitory co-stimulation molecules on ECs, such as PDL-1 and PDL2, can be maintained by a local delivery method of targeted rapamycin.29 Rapamycin pre-treatment of ECs has also been shown to reduce EC activation, proliferation of alloreactive memory T cells, and allow for the selective proliferation of graft protective T regulatory cells, in vitro and in vivo.3,29,30 In addition, these studies also established that rapamycin significantly reduces the secretion of IL-6.3 The ability of nano-based targeting of small interfering RNA (siRNA) to disrupt EC MHC expression and thereby reduce recruitment and activation in T cells in the setting of transplantation has also been successfully explored and bolsters the idea that pre-treatment in this setting may prevent long term dysfunction.2 Taken together, these data support the use of rapamycin encapsulated as a targeted nanotherapeutic micelle to abrogate transplant fibrosis and vasculopathy when used as a donor organ preservation pre-treatment strategy. In a recent study by Solhjou et al., novel controlled-release IL-6-containing nanoparticles were utilized as a pre-treatment method in experimental cardiac allografts to blunt early IRI injury and chronic rejection responses.31 In the novel studies presented here, we approach the problem of vasculopathy in a similar fashion in the preservation phase of transplantation prior to implantation of the allograft.
Conclusions
Here, for the first time, we demonstrate that delivery of rapamycin-encapsulated nanotherapy directly to allograft epithelial cells and ECs can prevent downstream organ dysfunction and improve cellular health in vitro while preventing vascular lesions and fibrosis of chronic rejection in vivo when compared to freely delivered rapamycin. In fact, high doses (1000 ng mL−1) of free rapamycin dissolved in the preservation solution resulted in an increase incidence of fatal tracheal dehiscence in our mouse model lung transplantation. We did not see this in the aortic interposition graft model, probably due to our use of a previously described cuffed anastomosis.18 The two separate models of transplantation provide proof-of-principle for rapamycin's clinical potential in donor organ pre-treatment. Therefore, inclusion of the FDA-approved rapamycin, in the form of a novel targeted nanotherapeutic micelle, can protect allografts from endothelial and epithelial activation, significantly reducing organ damage and potentially inducing a tolerogenic graft microenvironment.Conflicts of interest
Drs Carl Atkinson, Ann-Marie Broome and Satish Nadig are co-founders and Chief Scientific Officer, Chief Executive Officer, and Chief Medical Officer, respectively, of ToleRaM Nanotech, LLC. Drs Carl Atkinson, Ann-Marie Broome and Satish Nadig are inventors in patent Donor organ pre-treatment formulation, US2016/039315.Funding
These studies were supported by grants from the National Institutes of Health (NIH) National Institute of Biomedical Imaging and Biochemistry (NIBIB) K08 EB019495 (SNN), NIH Institutional Post-Doctoral Training Grant HL007260 (KP), AHA 17CPOST33671205 (CA, KP) and NIH Institutional Pre-Doctoral Training Grants 4T32 HL007260 and 5T32 GM008716 (DT). The South Carolina Clinical & Translational Research (SCTR) Institute supported portions of this work via NIH National Center for Advancing Translational Sciences (NCATS) grant number UL1 TR000062 (AMB and CA) and the National Natural Science Foundation of China 81700571 (QC). Supported in part by the Barclay Patterson Memorial Foundation (CA and SN). Supported in part by the Small Animal Imaging Shared Resource, Hollings Cancer Center, Medical University of South Carolina (P30 CA138313). The contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or NCATS.Author contributions
Concept and design of study: Carl Atkinson, Ann-Marie Broome, Satish Nadig. Acquisition of data: Peng Zhu, Suraj Dixit, Danh Tran, Kunal Patel, Yu-Lin Jiang, Scott Esckilsen, Kayla Miller, Grace Bazzle, Patterson Allen, Alfred Moore. Analysis and interpretation of data: Carl Atkinson, Ann-Marie Broome, Satish Nadig. Drafting manuscript: Carl Atkinson, Ann-Marie Broome, Satish Nadig. Revising manuscript critically for intellectual content: Carl Atkinson, Ann-Marie Broome, Satish Nadig. Approval of version of manuscript to be published: Carl Atkinson, Ann-Marie Broome, Satish Nadig.References
- H. Zhu, L. Yu, Y. He and B. Wang, Nonhuman primate models of type 1 diabetes mellitus for islet transplantation, J. Diabetes Res., 2014, 2014, 785948 CrossRef PubMed.
- J. Cui, L. Qin, J. Zhang, P. Abrahimi, H. Li and G. Li, et al. Ex vivo pretreatment of human vessels with siRNA nanoparticles provides protein silencing in endothelial cells, Nat. Commun., 2017, 8(1), 191 CrossRef PubMed.
- C. Wang, T. Yi, L. Qin, R. A. Maldonado, U. H. von Andrian and S. Kulkarni, et al. Rapamycin-treated human endothelial cells preferentially activate allogeneic regulatory T cells, J. Clin. Invest., 2013, 123(4), 1677–1693 CrossRef PubMed.
- W. Li, Q. Li, L. Qin, R. Ali, Y. Qyang and M. Tassabehji, et al. Rapamycin inhibits smooth muscle cell proliferation and obstructive arteriopathy attributable to elastin deficiency, Arterioscler., Thromb., Vasc. Biol., 2013, 33(5), 1028–1035 CrossRef PubMed.
- Y. Wang, Y. Bai, L. Qin, P. Zhang, T. Yi and S. A. Teesdale, et al. Interferon-gamma induces human vascular smooth muscle cell proliferation and intimal expansion by phosphatidylinositol 3-kinase dependent mammalian target of rapamycin raptor complex 1 activation, Circ. Res., 2007, 101(6), 560–569 CrossRef PubMed.
- O. Bamgbola, Metabolic consequences of modern immunosuppressive agents in solid organ transplantation, Ther. Adv. Endocrinol. Metab., 2016, 7(3), 110–127 CrossRef PubMed.
- K. Singh, L. Stempora, R. D. Harvey, A. D. Kirk, C. P. Larsen and B. R. Blazar, et al. Superiority of rapamycin over tacrolimus in preserving nonhuman primate Treg half-life and phenotype after adoptive transfer, Am. J. Transplant., 2014, 14(12), 2691–2703 CrossRef PubMed.
- J. Hester, A. Schiopu, S. N. Nadig and K. J. Wood, Low-dose rapamycin treatment increases the ability of human regulatory T cells to inhibit transplant arteriosclerosis in vivo, Am. J. Transplant., 2012, 12(8), 2008–2016 CrossRef PubMed.
- S. N. Nadig, S. K. Dixit, N. Levey, S. Esckilsen, K. Miller and W. Dennis, et al. Immunosuppressive nano-therapeutic micelles downregulate endothelial cell inflammation and immunogenicity, RSC Adv., 2015, 5(54), 43552–43562 RSC.
- N. Latchana, J. R. Peck, B. A. Whitson, M. L. Henry, E. A. Elkhammas and S. M. Black, Preservation solutions used during abdominal transplantation: Current status and outcomes, World J. Transplant., 2015, 5(4), 154–164 CrossRef PubMed.
- N. Anastasiou, S. Allen, R. Paniagua, A. Chester and M. Yacoub, Altered endothelial and smooth muscle cell reactivity caused by University of Wisconsin preservation solution in human saphenous vein, J. Vasc. Surg., 1997, 25(4), 713–721 CrossRef PubMed.
- M. L. Forrest, C. Y. Won, A. W. Malick and G. S. Kwon, In vitro release of the mTOR inhibitor rapamycin from poly(ethylene glycol)-b-poly(epsilon-caprolactone) micelles, J. Controlled Release, 2006, 110(2), 370–377 CrossRef PubMed.
- J. A. Yanez, M. L. Forrest, Y. Ohgami, G. S. Kwon and N. M. Davies, Pharmacometrics and delivery of novel nanoformulated PEG-b-poly(epsilon-caprolactone) micelles of rapamycin, Cancer Chemother. Pharmacol., 2008, 61(1), 133–144 CrossRef PubMed.
- G. W. Warren, M. A. Romano, M. R. Kudrimoti, M. E. Randall, R. C. McGarry and A. K. Singh, et al. Nicotinic modulation of therapeutic response in vitro and in vivo, Int. J. Cancer, 2012, 131(11), 2519–2527 CrossRef PubMed.
- M. Casiraghi, J. R. Tatreau, J. B. Abano, J. W. Blackwell, L. Watson and K. Burridge, et al. In vitro modeling of nonhypoxic cold ischemia-reperfusion simulating lung transplantation, J. Thorac. Cardiovasc. Surg., 2009, 138(3), 760–767 CrossRef PubMed.
- M. A. Khan, X. Jiang, G. Dhillon, J. Beilke, V. M. Holers and C. Atkinson, et al. CD4+ T cells and complement independently mediate graft ischemia in the rejection of mouse orthotopic tracheal transplants, Circ. Res., 2011, 109(11), 1290–1301 CrossRef PubMed.
- M. A. Khan, C. Maasch, A. Vater, S. Klussmann, J. Morser and L. L. Leung, et al. Targeting complement component 5a promotes vascular integrity and limits airway remodeling, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(15), 6061–6066 CrossRef PubMed.
- P. Zhu, S. Esckilsen, C. Atkinson, X. P. Chen and S. N. Nadig, A simplified cuff technique for abdominal aortic transplantation in mice, J. Surg. Res., 2016, 200(2), 707–713 CrossRef PubMed.
- G. Warnecke, A. Bushell, S. N. Nadig and K. J. Wood, Regulation of transplant arteriosclerosis by CD25+CD4+ T cells generated to alloantigen in vivo, Transplantation, 2007, 83(11), 1459–1465 CrossRef PubMed.
- T. Murakawa, M. M. Kerklo, M. R. Zamora, Y. Wei, R. G. Gill and P. M. Henson, et al. Simultaneous LFA-1 and CD40 ligand antagonism prevents airway remodeling in orthotopic airway transplantation: implications for the role of respiratory epithelium as a modulator of fibrosis, J. Immunol., 2005, 174(7), 3869–3879 CrossRef.
- S. N. Nadig, J. Wieckiewicz, D. C. Wu, G. Warnecke, W. Zhang and S. Luo, et al. In vivo prevention of transplant arteriosclerosis by ex vivo-expanded human regulatory T cells, Nat. Med., 2010, 16(7), 809–813 CrossRef PubMed.
- A. Dashkevich, A. Raissadati, S. O. Syrjala, G. Zarkada, M. A. Keranen and R. Tuuminen, et al. Ischemia-Reperfusion Injury Enhances Lymphatic Endothelial VEGFR3 and Rejection in Cardiac Allografts, Am. J. Transplant., 2016, 16(4), 1160–1172 CrossRef PubMed.
- L. Brasile, N. Henry and B. Stubenitsky, Underlying Mechanisms of Protection Involved in Immunocloak, Transplantation, 2017, 101(2), e49–e56 CrossRef PubMed.
- C. Zhang, Y. Niu, L. Yu, W. Lv, H. Xu and A. Abuduwufuer, et al. The role of epithelial-mesenchymal transition in the post-lung transplantation bronchiolitis obliterans, Journal of Cardiothoracic Surgery, 2017, 12(1), 119 CrossRef PubMed.
- L. R. Barbieri, D. D. Lourenco-Filho, E. R. Tavares, P. O. Carvalho, P. S. Gutierrez and R. C. Maranhao, et al. Influence of Drugs Carried in Lipid Nanoparticles in Coronary Disease of Rabbit Transplanted Heart, The Annals of Thoracic Surgery, 2017, 104(2), 577–583 CrossRef PubMed.
- A. I. Fiorelli, D. D. Lourenco-Filho, E. R. Tavares, P. O. Carvalho, A. F. Marques and P. S. Gutierrez, et al. Methotrexate associated to lipid core nanoparticles improves cardiac allograft vasculopathy and the inflammatory profile in a rabbit heart graft model, Braz. J. Med. Biol. Res., 2017, 50(10), e6225 Search PubMed.
- T. T. Pham, T. T. Nguyen, S. Pathak, S. Regmi, H. T. Nguyen and T. H. Tran, et al. Tissue adhesive FK506-loaded polymeric nanoparticles for multi-layered nano-shielding of pancreatic islets to enhance xenograft survival in a diabetic mouse model, Biomaterials, 2018, 154, 182–196 CrossRef PubMed.
- J. D. Fisher, A. P. Acharya and S. R. Little, Micro and nanoparticle drug delivery systems for preventing allotransplant rejection, Clin. Immunol., 2015, 160(1), 24–35 CrossRef PubMed.
- G. Tellides and J. S. Pober, Interferon-gamma axis in graft arteriosclerosis, Circ. Res., 2007, 100(5), 622–632 CrossRef PubMed.
- B. Fogal, T. Yi, C. Wang, D. A. Rao, A. Lebastchi and S. Kulkarni, et al. Neutralizing IL-6 reduces human arterial allograft rejection by allowing emergence of CD161+ CD4+ regulatory T cells, J. Immunol., 2011, 187(12), 6268–6280 CrossRef PubMed.
- Z. Solhjou, M. Uehara, B. Bahmani, O. H. Maarouf, T. Ichimura and C. R. Brooks, et al. Novel Application of Localized Nanodelivery of Anti-Interleukin-6 Protects Organ Transplant From Ischemia-Reperfusion Injuries, Am. J. Transplant., 2017, 17(9), 2326–2337 CrossRef PubMed.
Footnotes |
| † These authors are members of the South Carolina Investigators in Transplantation (SCIT) Consortium. |
| ‡ These authors are members of the Hollings Cancer Center (HCC) Small Animal Imaging Center. |
| This journal is © The Royal Society of Chemistry 2018 |