
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding GPCR recognition and folding from NMR studies of fragments
Jacopo Marino
 a,
Reto Walser
db,
Martin Poms
dc and
Oliver Zerbe
*d
a,
Reto Walser
db,
Martin Poms
dc and
Oliver Zerbe
*d
aLaboratory of Biomolecular Research, Paul Scherrer Institute, Villigen, Switzerland
bAstex Pharmaceuticals, 436 Cambridge Science Park, Milton Road, Cambridge, CB4 0QA, UK
cDepartment of Neurology, Children's Hospital Zurich, Steinwiesstrasse 75, CH-8032 Zurich, Switzerland
dDepartment of Chemistry, University of Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland. E-mail: oliver.zerbe@chem.uzh.ch; Fax: +41-44-6356882; Tel: +41-44-6354263
First published on 9th March 2018
Abstract
Cotranslational protein folding is a vectorial process, and for membrane proteins, N-terminal helical segments are the first that become available for membrane insertion. While structures of many G-protein coupled receptors (GPCRs) in various states have been determined, the details of their folding pathways are largely unknown. The seven transmembrane (TM) helices of GPCRs often contain polar residues within the hydrophobic core, and some of the helices in isolation are predicted to be only marginally stable in a membrane environment. Here we review our efforts to describe how marginally hydrophobic TM helices of GPCRs integrate into the membrane in the absence of all compensating interhelical contacts, ideally capturing early biogenesis events. To this end, we use truncated GPCRs, here referred to as fragments. We present data from the human Y4 and the yeast Ste2p receptors in detergent micelles derived from solution NMR techniques. We find that the secondary structure in the fragments is similar to corresponding parts of the entire receptors. However, uncompensated polar or charged residues destabilize the helices, and prevent proper integration into the lipid bilayer, in agreement with the biophysical scales from Wimley and White for the partitioning of amino acids into the membrane-interior. We observe that the stability and integration of single TM helices is improved by adding neighboring helices. We describe a topology study, in which all possible forms of the Y4 receptor were made so that the entire receptor is truncated from the N-terminus by one TM helix at a time. We discover that proteins with an increasing number of helices assume a more defined topology. In a parallel study, we focused on the role of extracellular loops in ligand recognition. We demonstrate that transferring all loops of the human Y1 receptor onto the E. coli outer membrane protein OmpA in a suitable topology results in a chimeric receptor that displays, albeit reduced, affinity and specificity for the cognate ligand. Our data indicate that not all TM helices will spontaneously insert into the helix, and we suggest that at least for some GPCRs, N-terminal segments might remain associated with the translocon until their interacting partners are biosynthesized.
 Jacopo Marino | Jacopo Marino is working at the Laboratory of Biomolecular Research of the Paul Scherrer Institute in Switzerland, using cryo-electron microscopy to reveal the architecture of membrane protein complexes. Jacopo obtained his PhD from the University of Zurich working with Oliver Zerbe. After his PhD, Jacopo moved to the LMU-Munich to work as a postdoc in the laboratory of Prof. Beckmann, where he further investigated how small protein domains fold cotranslationally. Jacopo is currently combining his experience in molecular biology and cryo-EM with the aim of becoming an independent group leader. |
 Reto Walser | Reto Walser studied biochemistry and biophysics at the University of Basel and at the ETH Zurich. He then joined Oliver Zerbe's group at the University of Zurich for a PhD on solution NMR applied to membrane proteins. After postdoctoral projects at the Institute of Physical Chemistry of the University of Zurich and at AstraZeneca (UK), he joined Astex Pharmaceuticals in Cambridge, UK, where he is now the head of biomolecular NMR. His interests include NMR methods for enabling fragment-based drug discovery and the physico-chemical properties underlying protein-ligand interactions. |
 Martin Poms | Martin Poms is currently a postdoctoral research associate at the Children's Hospital Zurich (Switzerland). He studied chemistry at the Graz University of Technology (Austria), where he obtained his Masters in 2009. In 2015 he received his PhD from the University of Zurich with Oliver Zerbe, investigating membrane protein structures and folding using NMR spectroscopy. His current research interests include small-molecule profiling of inborn errors in metabolism, biomarker identification and the development of analytical methods in small-molecule detection. |
 Oliver Zerbe | Prof. Oliver Zerbe studied chemistry in Hamburg, Germany. He worked on NMR methodology applied to peptides in his PhD thesis at the University of Zurich with von Philipsborn. After a postdoctoral stay in the lab of Nobel laureate Kurt Wüthrich he became head of the NMR facilities, first in the Institute of Pharmaceutical Sciences of the ETH with Gerd Folkers, then independently at the University of Zurich. In his research he utilizes solution NMR techniques and other biophysical methods to study membrane proteins, in particular GPCRs, as well as the design and folding of repeat proteins. Moreover, he is interested in the structure of metallothioneins. |
Introduction
G-protein coupled receptors (GPCRs) constitute a very important class of integral membrane proteins that help transmit a signal from the outside to the inside of cells. Around 35% of all commercial drugs are believed to act via binding to GPCRs.1 A network of interactions between the seven transmembrane (TM) helices, formed by hydrogen bonds, salt-bridges and hydrophobic contacts confers plasticity upon activation, thus allowing GPCRs to respond to different ligands and bind different intracellular effector proteins.2 The characterization of subtle structural changes upon GPCR activation is crucial to developing ligands that activate specific intracellular pathways exclusively and minimize side effects.3While the first low-resolution data emerged from cryo-electron microscopy in 1993![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4 the scientific community had to wait until 2000 for the first crystal structure to appear, the structure of bovine rhodopsin.5 Since then we have witnessed the publication of new structures at an ever-accelerating pace culminating in the Nobel prize in chemistry awarded to Brian Kobilka6 and Bob Lefkowitz7 in 2012. The major breakthroughs in this area of structural biology have recently been reviewed by Grisshammer.8
4 the scientific community had to wait until 2000 for the first crystal structure to appear, the structure of bovine rhodopsin.5 Since then we have witnessed the publication of new structures at an ever-accelerating pace culminating in the Nobel prize in chemistry awarded to Brian Kobilka6 and Bob Lefkowitz7 in 2012. The major breakthroughs in this area of structural biology have recently been reviewed by Grisshammer.8
Early studies of folding of GPCRs and GPCR-like proteins comprise the seminal work of the Khorana group that demonstrated that bacteriorhodopsin (bR) can be refolded from its fully denatured state.9 While refolding of most GPCRs from the denatured state is difficult but not impossible, no structure of a refolded GPCR has been published so far. Most of the folding studies of GPCRs rather focused on unfolding them in chaotropic agents10 using a number of biophysical or computational11 techniques to characterize the folding intermediates and to detect folding cores. However, most of the experimental techniques cannot deliver dynamical information at atomic resolution with the notable exception of single-molecule techniques such as atomic force microscopy (AFM). Those can be used to map overall conformal preferences of membrane proteins, even if they do not adopt a unique topology.12–14
The potential of investigating dynamical features of GPCRs by NMR has been recognized early on, but studies of GPCRs by NMR are largely hampered by technical issues.15 The seminal paper by the Nietlispach group reporting on sensory rhodopsin16 and the structure of proteorhodopsin17 have indicated that solution NMR in principle is capable of determining the structures of 7-TM membrane proteins. In addition, GPCRs labeled with only certain amino acid types18–22 or receptors covalently linked to fluorine moieties23,24 have been used to investigate aspects of receptor activation.
In this review we summarize our work on the use of truncated GPCRs, here referred to as “fragments”, to study interhelical contact formation in neighboring TM helices, and ligand recognition by extracellular loops and the N-terminal domain. The different approaches we followed for using fragments are depicted in Fig. 1.
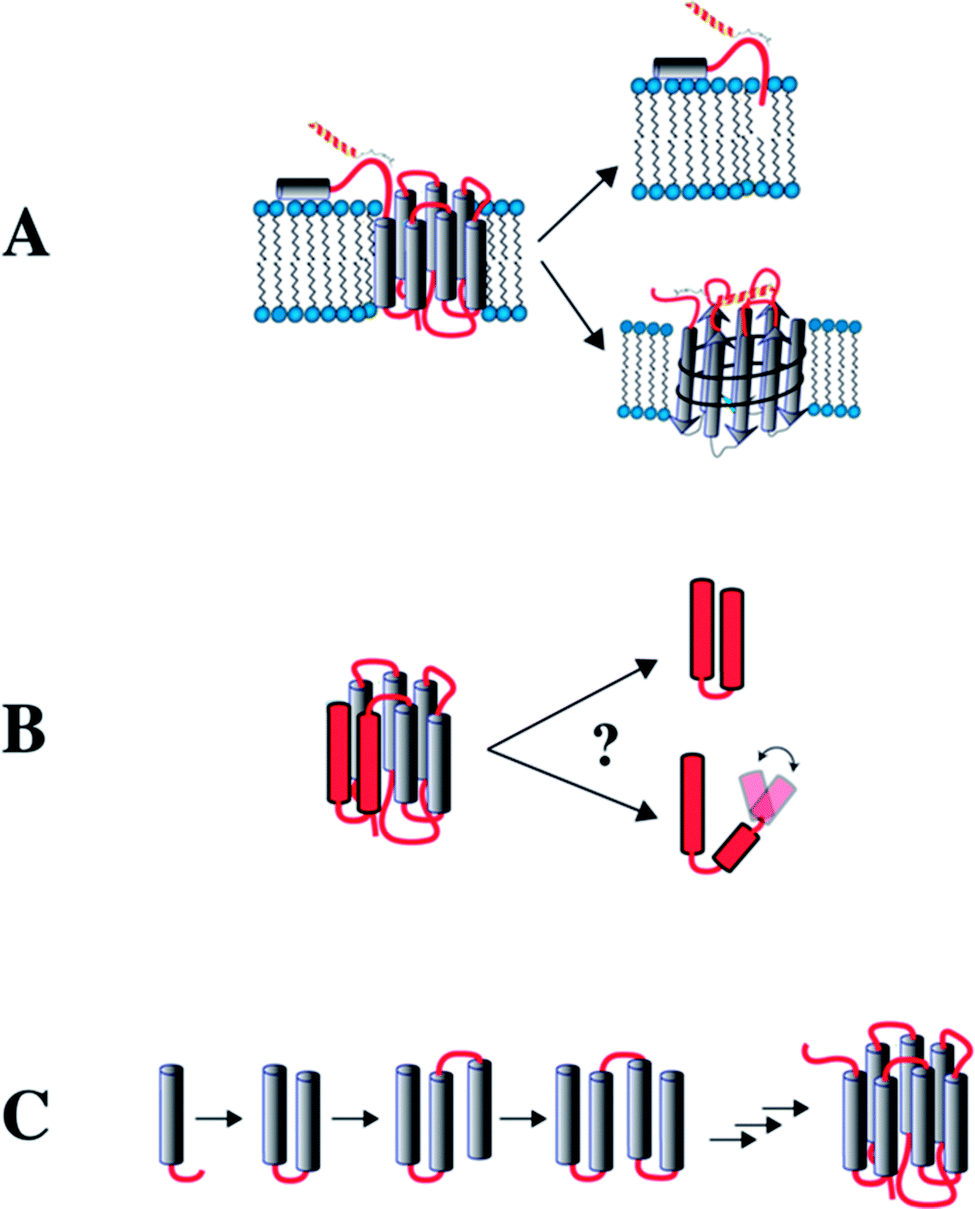 | ||
| Fig. 1 Potential approaches to study GPCRs from fragments. (A) Recognition of extracellular (EC) domains. The EC N-terminal domain is studied in isolation. Alternatively, the EC loops are grafted onto a β-barrel protein (OmpA) to study binding by NPY peptide ligands. (B) Formation of interhelical contacts studied in receptors that have been dissected into 2-TM fragments. (C) NMR is used to study fragments of increasing length to infer interhelical contact formation and stability of the TM helices during biogenesis in a lipid environment. | ||
We focus on examples from two GPCR families: the human Y receptors and the yeast mating pheromone receptor Ste2p. The Y receptor family consists of four members (Y1, Y2, Y4 and Y5), which all are class A receptors that bind to peptides of the NPY family. The Y receptors mediate vasoconstriction, stimulation of food intake, intestinal functions, regulation of circadian rhythms, and the release of pituitary sex hormones.25 We began by studying the binding mechanisms of the NPY ligands to the Y receptors. We discovered that binding is orchestrated via a two-step mechanism, which includes first association to the cellular membrane and a second recognition by the extracellular N-terminal domain and by the extracellular loops of the GPCRs26 following the membrane compartment model initially postulated by Schwyzer.27,28 For these reasons, the peptides of the NPY family represent an excellent class of ligands whose binding to membranes or membrane mimetics can be studied in great detail by NMR. We further used the Y4 receptor to study interhelical contact formation in neighboring TM helices. The S. cerevisiae Ste2p receptor binds the tridecapeptide α-factor mating pheromone, resulting in growth arrest and gene regulation in preparation for mating.29 Studies on this receptor were carried out in collaboration with the groups of Naider and Becker, and largely concern the question of how uncompensated polar residues at central helix position influence protein topology.
The membrane compartment model indicates that the extracellular N-terminal domains or the extracellular loops of the Y receptors may interact with their ligands. For this reason, we produced the peptides corresponding to the N terminal domains of the Y-receptors and studied by NMR their interaction with their ligands (Fig. 1A). We then created a model for the extracellular loops of the Y1 receptor by grafting the latter in a topologically related fashion onto a well-behaved membrane protein scaffold derived from the bacterial outer membrane protein OmpA (Fig. 1A).30,31 This allowed us to study recognition of the peptide ligands by the loops.32,33 Subsequently, we made 2–3 TM fragments to study the importance of inter-helical contacts (Fig. 1B), and complemented this approach by studying folding of a series of overlapping N-terminal fragments of increasing length (Fig. 1C) by NMR. We further complemented the NMR data with studies on the orientation of these fragments in the inner membrane of E. coli. Finally, we briefly mention the use of complementary fragments to make a mimic of a segmentally labeled entire GPCR.
General remarks on studying protein fragments
Does it make much sense to look at protein fragments? This question is highly disputed. The seminal work from the Kay group on the study of the entire proteasome by solution NMR using a divide-and-conquer approach34 showed that such an approach can elegantly solve many spectroscopic issues of a system of such size. However, in general the answer for soluble proteins is “no” because dissecting a protein within domain borders will usually result in unstructured and insoluble aggregates. For certain membrane proteins, however, this approach might still work. Here, secondary structure formation is promoted by the hydrophobic environment, because hydrogen bonds are formed to shield the polar peptide bond from making unfavorable contacts to lipids.35,36 The successful design of split receptors of rhodopsin37 and the α-factor receptor Ste2p38 indicated that receptor fragments can complement each other, restoring the functionality of the full-length receptor. The first structure determination of a GPCR fragment by NMR39 was encouraged by the finding that cytoplasmic fragments of rhodopsin inhibit the interaction between rhodopsin and its G-protein transducin.40 However, the exact structural architecture is often not maintained in GPCR fragments, and hence we feel that the study of fragment structures is not feasible in order to obtain an atomistic picture of the entire receptor. Instead, we used GPCR fragments to address questions of ligand recognition and folding that would be difficult to address using full-length receptors.The two-stage membrane-protein folding model proposed by Popot and Engelman postulates that segments of membrane proteins form helical structures upon partitioning into the water–membrane interface.41 These helices subsequently insert into the hydrophobic core of the membrane and diffuse within the bilayer until contacts with other helices are made eventually leading to the assembly of the helical bundle. If secondary structure in membrane environments is predominantly determined by sequence, and does not depend on the formation of tertiary contacts, it should be retained in fragments. As described below in more detail, we usually observe good agreement between the location of helical segments and structural models of our GPCRs, an indication that formation of secondary structure of membrane proteins is indeed primarily encoded in the immediately neighboring amino acid sequence.
Is tertiary structure correctly retained in fragments? A number of features were proposed to be important for the formation of inter-helical contacts.42–45 Those are for example shape-complementary amino acids, e.g. the knobs-in-hole arrangement in GXXXG motifs.46,47 Another way to mediate helical contacts is through formation of aromatic sidechain interactions48 or via formation of hydrogen bonds.49 The latter requires the presence of polar residues in sequence locations where they would not be expected based on a match of hydrophobicity with the surrounding lipid environment. It is obvious that exposure of polar residues to lipids will hamper proper integration of the corresponding stretch into the membrane, and will additionally destabilize secondary structure.50 The location of polar residues situated within the membrane is important since it provides specificity for interhelical contacts, which should be taken into account by selecting the fragment such that most of the polar residues find their interaction partners within the same fragment if possible. On the other hand, looking at fragments also offers the possibility to study the behavior of an N-terminal segment of the protein in the absence of the remainder of the protein, ideally mimicking what happens during early biogenesis events within the lipid bilayer.
Synthetic aspects
Biosynthesis of GPCR fragments
E. coli has been the host of choice for obtaining proteins for NMR studies because of the possibility to introduce isotope labels in almost all flavors.Experience from our and other laboratories indicates that it is possible to generate milligram quantities of purified GPCR fragments from 1–2 L of E. coli culture, even when growing cultures with minimal media in deuterated water.50–57 We have observed that some GPCR fragments are well produced in E. coli, while others require the use of an N-terminal fusion partner to increase expression. Among the N-terminal fusion proteins that we have employed to increase expression are the ΔTrp-leader sequence58 or Mistic.59
During protein biogenesis in E. coli, GPCR fragments are intrinsically prone to aggregation due to their hydrophobic nature and lack of a recognition sequence for the Sec translocon. Thus, they accumulate in the form of inclusion bodies. However, inclusion bodies can be solubilized with the aid of organic solvents such as trifluoroacetic acid (TFA),60 or detergents such as SDS or fos-choline-12,51 among other strategies followed in our group to solubilize insoluble proteins.61,62
Once GPCR fragments are solubilized from inclusion bodies, we performed a chromatographic separation in presence of denaturants, such as reverse-phase column or ion metal affinity chromatography (IMAC), followed by dialysis against water to remove the denaturant, and lyophilization. Purity and the correct mass of the protein obtained is verified by mass-spectrometry. This procedure offers the unique advantage that the lyophilized material can be solubilized in the required volume of buffer containing detergent, a prerequisite to obtain reproducible NMR measurements. Furthermore, solubilization of lyophilized material into small volumes of buffers minimizes the use of expensive deuterated detergents.
We have experienced that some GPCRs fragments could not be efficiently re-solubilized after lyophilization. This prompted us to investigate the expression into the inner membrane of E. coli. GPCRs lack N-terminal signal sequences for insertion into the membrane, but expression of functional GPCRs in E. coli sometimes can be achieved by using N-terminal fusion partners such as the maltose binding protein (MBP),63 or Mistic.59 The use of N-terminal fusions proved to be useful for GPCR fragments, and allowed us to investigate how the transmembrane helices of the fragments are oriented in respect to the lipid bilayer of the E. coli inner membrane (vide infra).
Recognition of GPCRs by their ligands
Our group studied members of the NPY family, ligands for the Y-receptor GPCRs, using high-resolution NMR both in solution and in the membrane-bound state.64–67 Based on these studies we proposed a model for the binding of these hormones to their cognate receptors,26 which suggests that initial binding of the ligands to the membrane constitutes an integral part of the receptor recognition process.27,68For larger ligands, and in particular in case of peptide ligands, photoaffinity-labeling studies revealed that contact points between receptors and their ligands can be assigned to the extracellular face of GPCRs, which is comprised of an N-terminal domain and three extracellular loops (ECLs) of variable size.69–71 To investigate this aspect we have studied the interactions of peptides of the NPY family with isolated N-terminal domains72 or with a chimeric receptor in which all extracellular loops as well as the N-terminal domain were grafted onto a β-barrel scaffold, the membrane protein OmpA.32,33
The role of the N-terminal extracellular part
We initially investigated conformational preferences of the extracellular N-terminal domains of all Y receptors.72 While they are largely unfolded, in case of the Y4 receptor an amphiphilic helix is formed, which is anchored on the micelle surface, connected via a long and flexible loop to TM1 (Fig. 2). Interestingly, the ligand pancreatic polypeptide (PP), another member of the NPY family of neurohormones, binds with a 50 μM affinity to that loop as determined by surface-plasmon resonance, largely via electrostatic interactions.73 In a first step, ligands associate with the membrane surface, and diffuse laterally.26 In proximity to the receptor a weak interaction of PP with the N-terminal domain helps transferring the peptide from the membrane-bound state to the receptor binding-pocket following a fly-casting mechanism (Fig. 2).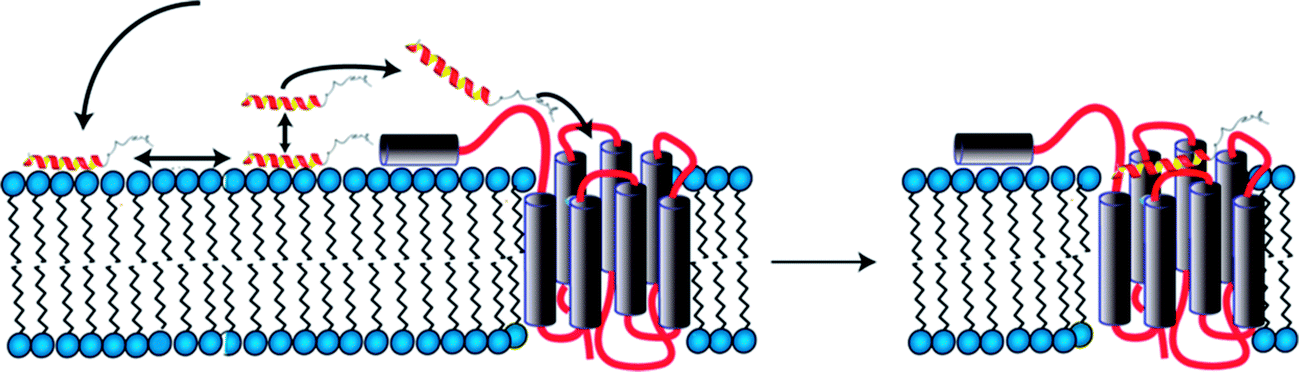 | ||
| Fig. 2 Model for binding of PP to the Y4 receptor comprising association with the membrane surface (left), formation of transient contacts to the N-terminal domain (left) and diffusion into the orthosteric binding pocket (right). | ||
The role of extracellular loops
Binding affinities to isolated receptor loops are often in the μM range74 while binding affinities to full-length receptors commonly are in the nM range (see http://gpcrdb.org/interaction/ for an overview of ligand binding data). The cumulative effects of two or more μM binding sites present in two separate loops can explain the higher binding-affinities for the entire receptor, amongst other factors.Since formation of proper tertiary structure per se is questionable in fragments, it is highly unlikely that the loop conformations in isolated TM fragments will be meaningful. Grafting the loops onto a suitable scaffold could allow for the simultaneous display of several (ideally all) binding epitopes of a GPCR in approximately correct relative orientations and could hence serve as a suitable GPRC-mimic for biophysical or structural studies. We therefore selected a scaffold that can accommodate at least three extracellular loops of the Y1 receptor, can easily be biosynthesized and handled, resides in the membrane, and also can be characterized by solution NMR.
In the resulting chimeric receptor, we grafted the loops of the Y1 receptor onto the β-barrel scaffold derived from the E. coli outer membrane protein A (OmpA) by replacing three of its four loops at one side of the barrel by the Y1 receptor extracellular loop sequences (Fig. 3).32,33 The three loops can be transferred in various arrangements onto the scaffold. We ranked the different topomers according to how similar distances of loop-anchoring residues are with respect to known GPCR structures and chose to biosynthesize four candidates that most closely resemble the native receptors. All of those constructs expressed well and could be refolded. Using chemical shift mapping and saturation-transfer NMR techniques we demonstrated that the chimeric receptor recognizes the native ligand NPY in a specific manner.32 We have also transferred the extracellular domain onto the remaining accessible β-barrel scaffold loop acceptor position, but could not detect an increase in binding affinity.33 The affinity between these chimeric Y1 receptors and NPY are in the micromolar range, which is 2–3 orders of magnitude weaker than the interaction between NPY and the native Y1 receptor. This discrepancy can be attributed to differences in the topology of the chosen template, in particular in different relative locations of the loop anchoring points. Alternatively, X-ray structures of GPCRs with peptide ligands,75 unknown at the outset of our study, later revealed that additional interactions formed between the ligand and TM residues are involved in ligand binding.
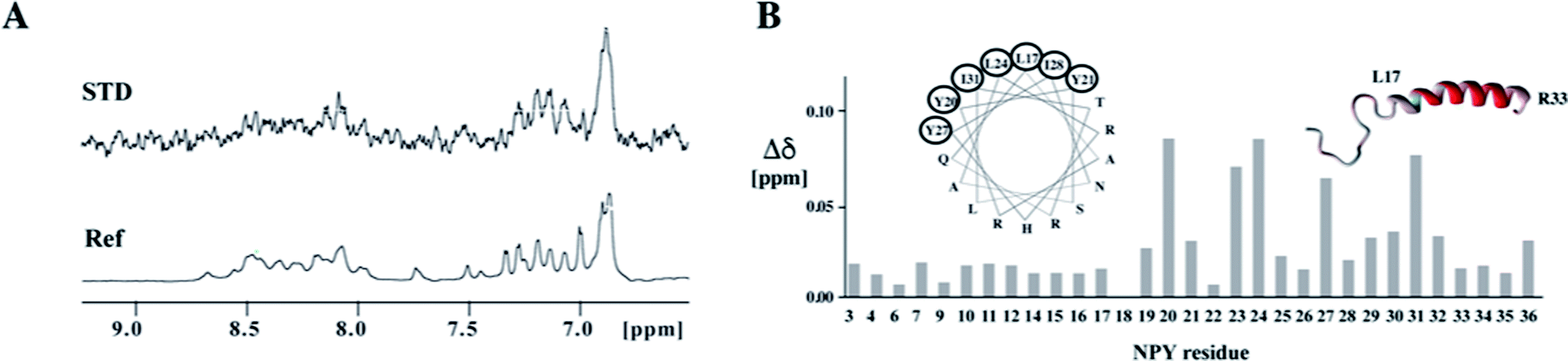 | ||
| Fig. 3 Binding of the ligand peptide NPY to the chimeric model of the Y1 receptor. (A) Ligand binding studies by saturation transfer difference (STD). The reference spectrum is depicted at the bottom. The strong STD signal at ∼6.9 ppm stems from the aromatic protons of Y27. (B) Chemical shift perturbations in NPY upon binding to the chimeric receptor. Residues for which amide resonances are shifted to the largest extent are encircled in the helical wheel presentation and are colored in red in the ribbon representation of NPY. | ||
Grafting loops of GPCRs onto soluble scaffolds has been successfully demonstrated in case of the RXFP1 receptor76,77 using the B1 immunoglobulin binding domain of the streptococcal protein G (GB1) as the scaffold. Similarly, the N terminal domain and the third extracellular loop of the PTH receptor were grafted onto the four-helix bundle protein TM1526 from the thermophilic archaea bacteria Thermotoga maritime.78 Last but not least it was also demonstrated that a polypeptide, in which the extracellular loop sequences of the CXCR4 receptor were connected via appropriate spacers recognizes gp120 with good affinity.79,80
Folding of GPCRs
A study of overlapping TM fragments from the Ste2p receptor
Cotranslational insertion of membrane proteins into the lipid bilayer is a vectorial process that starts from the N-terminus of the nascent chain. When studying folding of GPCRs, the use of N-terminal fragments presents a conceptual advantage over investigating the entire receptors, because C-terminal segments of the receptor are not present when the first N-terminal segments have been made and are ready for insertion into the membrane. Since we suspected that the TMs mutually stabilize each other, we set out to investigate a series of N-terminal fragments derived from the yeast GPCR Ste2p of increasing size that always start with TM1 (Fig. 4).81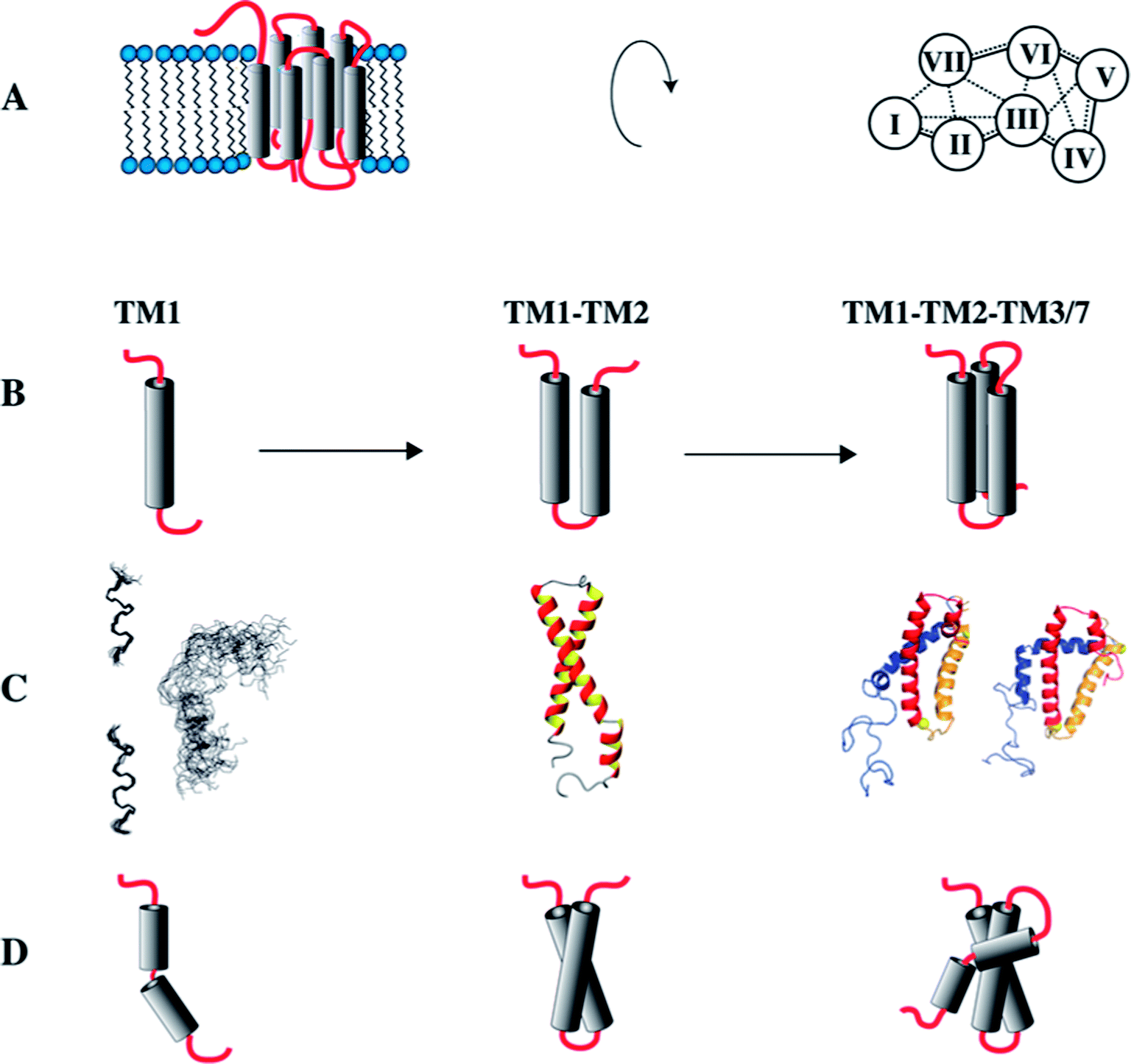 | ||
| Fig. 4 Study of C-terminally truncated forms of the Ste2p receptor. (A) Schematic representation of the Ste2p receptor (left) and inter-helical contacts (right). (B) Schematic representations of TM1, TM12 and TM123/TM127. (C) Experimental (NMR) structures of TM1, when individually superimposing individual conformers of the N- and C-terminal halves of TM1 separately (left), or when superimposing all residues (right). Other structures present conformers of TM12 or TM127 (two different conformers depicted). (D) Schematic representations of the experimental structures. | ||
Initially, we determined the structure of TM1 in LPPG/DPC mixed micelles. Instead of one continuous helix our NMR analysis revealed that this fragment is composed of two separate, rather well-defined short helices comprising residues Thr48–Phe55 and Ala61–Ile71 (Fig. 4C). These two helical segments are spaced by the amino acid sequence GSRVG. The orientation of the two short helices relative to each other was poorly defined.
When adding residues from the second TM, we detected interhelical contacts between TM1 and TM2 via NOEs between methyl groups, and TM1 now formed a continuous helix. This observation indicates that the presence of TM2 stabilizes secondary structure (around the GXXXG motif) in TM1 when compared to the isolated TM1, resulting in a helical hairpin for TM12 (Fig. 4C and D).
Unfortunately, the fragment TM123 is rather unstable and tends to aggregate. However, backbone assignments of TM123 were still possible, and both deuterium amide exchange and attenuation from soluble spinlabels indicated that there is water-access to the central part of TM3. This indicates that TM3 does not stably pack against TM1 and/or TM2. In contrast, the construct TM127, in which TM7 is artificially covalently linked to the loop of TM2, proved to be much more stable. Assuming that TM7 packs against TM1 and TM2 in the entire receptor,82 this fragment allowed us to investigate the formation of inter-helical contacts. Spectroscopic properties of TM1 and TM2 in TM127 changed only minimally, indicating that the helical hairpin is not significantly altered upon addition of TM7. However, TM7 similarly as TM3 does not stably pack against TM1 or TM2, but displays motions around an internal flexible hinge comprised by the central Leu-Pro-Leu tripeptide motif. In contrast to TM3, however, TM7 appears to be better integrated into the hydrophobic core, as we did not observe access of water to TM7.
Two principles emerged from that study that are particularly worth mentioning: (i) the stability of secondary and tertiary structure depends to a large extent on the presence of polar or charged residues in regions that are placed within the membrane interior. (ii) Fragments in general do not stably insert into the membrane.
We like to briefly mention that we started our work on fragments of the Ste2p receptor with TM7, using a construct that also comprises 40 residues from the C-terminal intracellular domain.83 The protein was expressed and purified in the lab of Fred Naider, and we determined the structure in DPC micelles. Similarly to TM1, the NMR data revealed that TM7 is not stably folded but rather exists in form of two helical stretches interrupted around a central Pro residue. The two helix portions are not stably anchored in the hydrophobic core of the micelles but transiently buried.
All NMR studies described above are performed using detergent micelles as membrane mimetics, and these have been used also for many crystallographic studies of membrane proteins. Detergent micelles, however, do not perfectly reflect planar bilayers as encountered in biological membranes nor do they contain non-lipid components such as other membrane proteins (integral or peripheral) or glycolipids. To obtain data on partitioning into real biological membranes we have collaborated with the group of Ismael Mingarro from Alicante University. His group performed assays on liver microsomes, relying on the absence or presence of glycosylation to probe for the periplasmic location of the C-terminus of the fragments.81 The conclusion from that work was that TM1 is not integrated into the membrane but TM12 and TM123 or TM127 mostly assumed native-like topology. A peptide corresponding to the more hydrophobic TM2 did insert into the membrane, and replacing Arg-58 at the center of TM1 by Leu or Ala also resulted in insertion. These studies indicate a pivotal role of uncompensated polar or charged residues within central locations of TM helices for folding and membrane-integration.
The importance of interhelical contacts
51 to obtain a series of overlapping polypeptides that would span the entire Y4 receptor. We managed to obtain close-to-complete backbone chemical shift assignments for most residues, both from loops as well as from the TM helices, with the notable exception of most residues from TM3 and TM4. Secondary chemical shifts of assigned TM residues indicate that the secondary structure in the putative TM portions is correct in the 2-TM fragments. We suspect that resonances from residues TM3 and TM4 were broadened through conformational exchange, reflecting the fact that TM helices often do not pack stably against each other in fragments. However, conformational broadening is also observed on entire receptors and the intrinsic instability is of importance for the mode of receptor activation.Interestingly, in contrast to the corresponding fragment from the Ste2p receptor the structure of TM12 of the Y4 receptor did not show tertiary contacts.50 Secondary structure is largely formed, however, and interrupts occur close to Gly, Pro or polar residues. A comparison of these two structures is depicted in Fig. 5. Please note the much more frequent occurrence of polar residues in TM1 or TM2 of the Y4 receptor when compared to the Ste2p receptor.
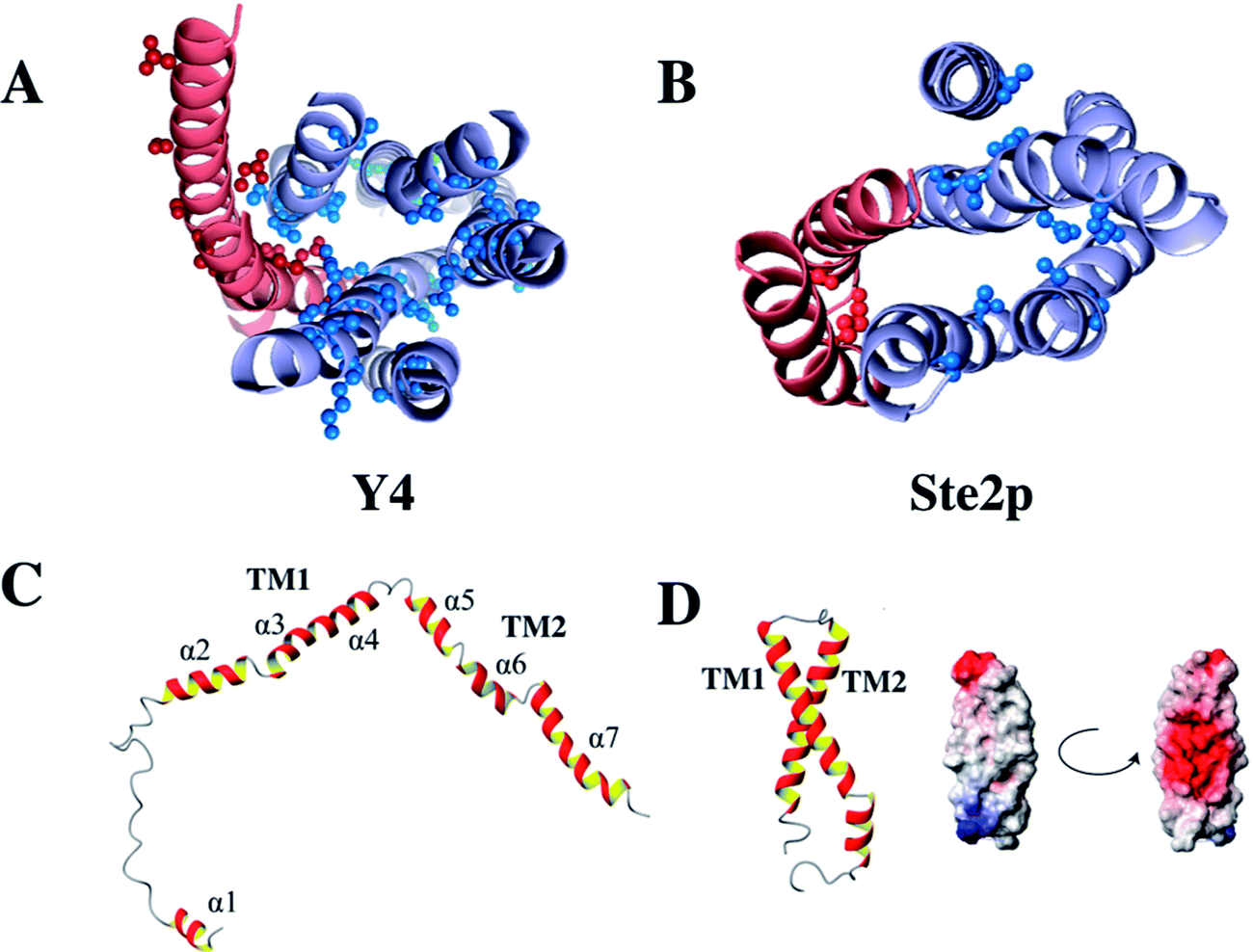 | ||
| Fig. 5 Location of polar residues in TM helices in the Y4 (left) and Ste2p (right) receptors. Models of the Y4 (A) and Ste2p (B) receptors. Atoms of side-chains of polar residues in TM helices at least 6 residues away from the helix ends are depicted as spheres. TM1 and TM2 are colored in red, TM3–TM7 in light blue. (C) Single NMR conformers of TM12 from the Y4, and (D) the Ste2p receptors along with the surface potential of TM12 of Ste2p. Positively and negatively charged surfaces are color coded in red and blue, respectively. | ||
In principle, one could try to construct an entire receptor from two complementary fragments. If such a fragment complex would be capable of still binding the ligand, the method would facilitate the NMR resonance assignment tremendously. Seminal work by the Khorana lab, which reconstituted active bacteriorhodopsin from complementary fragments, indicated that this should indeed be feasible.85 Similarly Dumont could reconstitute the Ste2p receptor from several pairs of complementary fragments.38 Note that in the work by the Dumont group the two fragments were co-expressed, and the Khorana group used cleavage of reconstituted bacteriorhodopsin to generate the fragments. We have tried to mix separately expressed fragments of the Ste2p receptor, e.g. TM1286 with TM34567,87 but so far failed to develop protocols to form protein complexes from fragments individually dissolved in detergents.
Folding of GPCRs in vivo
To compare the data obtained in vitro by NMR with the topology of fragments in a cellular membrane environment, and to possibly determine whether the lack of interhelical contacts that we observed for TM12 of the Y4 receptor was influenced by the detergent used for NMR (Fig. 5), we have performed a topology analysis by truncating the entire receptor by one helix at a time, starting from both the N-terminus and the C-terminus.89 We used GFP as a reporter for cytoplasmic and alkaline phosphatase (PhoA) as a reporter for periplasmic localization.88 A schematic of the approach is presented in Fig. 6A and B.
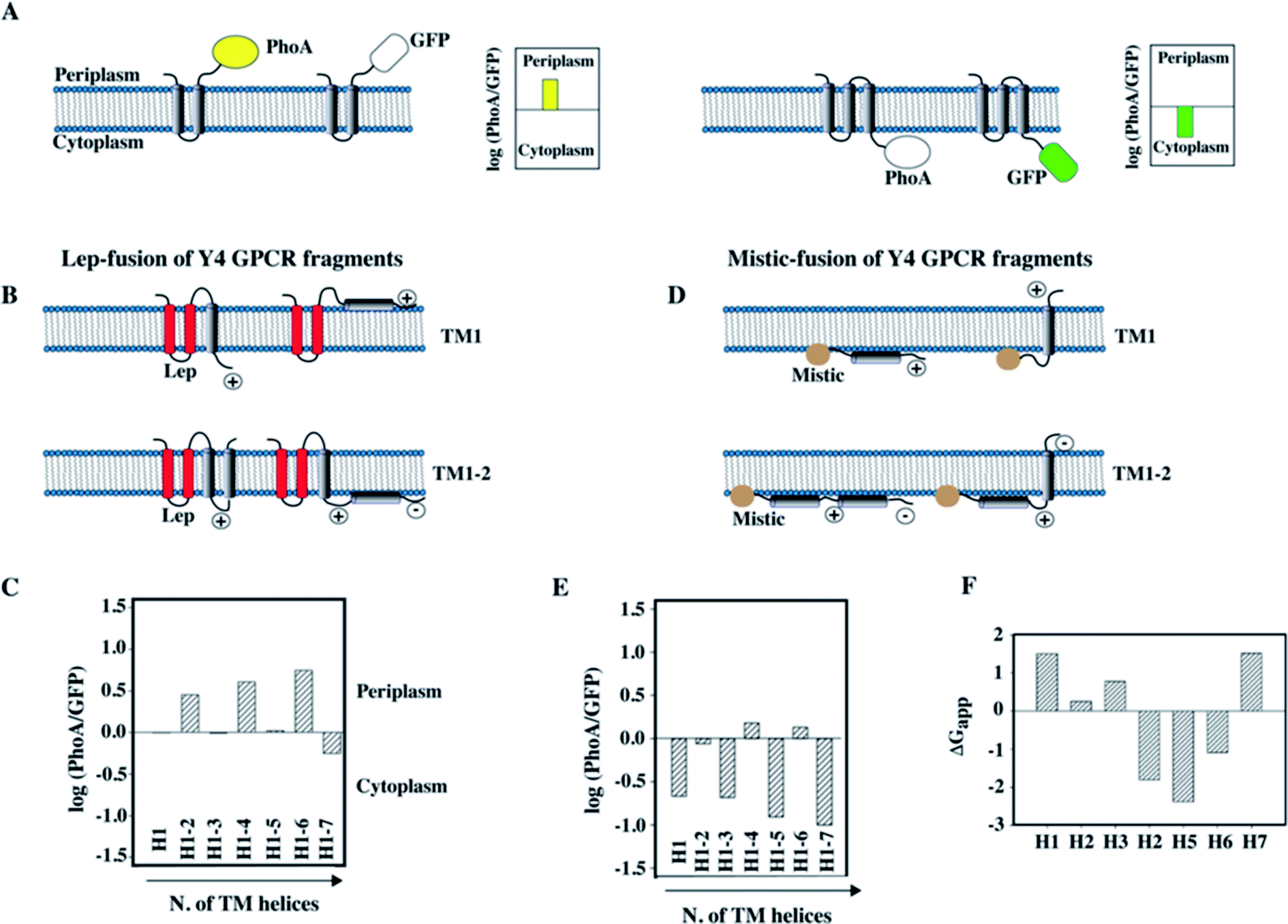 | ||
| Fig. 6 Topology analysis of Y4 GPCR fragments of increasing length into the inner membrane of E. coli. (A) Schematic representation of the assay used to study the topology of membrane insertion. When the C-terminus of a membrane protein is located in the periplasm, fusion to alkaline phosphatase (PhoA) allows detection of enzymatic activity. In case of cytoplasmic location of the C-terminus, GFP fluorescence can be measured. (B) Schematic representation of the Lep-fusion to TM1 and TM12 of the Y4 receptor and their possible topologies in the E. coli inner membrane, considering the distribution of positive and negative charges in the loops and the occurrence of polar or charged residues within the TM helices. (C) Experimental results of the topology assay of the Lep-fusions constructs, reporting the Y4 fragments of increasing length starting from the N-terminus of the Y4 receptor. (D) Schematic representation of the Mistic-fusion to TM1 and TM12 of the Y4 receptor, and possible topologies considering the distribution of positive and negative charges in the loops. (E) Experimental results of the topology assay for fragments of the Y4 receptor fused to Mistic. (F) Free energies for partitioning the entire TM peptide into the membrane using the scale from Wimley and White.92,93 | ||
Since the N-terminus is likely either in the periplasmic or in the cytoplasmic compartments for the N-terminally truncated constructs, depending on how many helices were removed, we have used two different N-terminal fusion proteins to perform the study: the transmembrane region of the bacterial peptidase Lep was used to ensure proper periplasmic orientation,90 while Mistic was used to ensure cytoplasmic orientation (Fig. 6B and D).
The information acquired by combining the results from the GFP and PhoA reporters allowed quantifying the fraction of expressed protein that folds in a topology that resembles the organization in the entire receptor. Interestingly, we could observe that N-terminal fragments do not acquire a unique topology, however, the fraction displaying the correct localization of the C-terminus increased with the number of helices that were added. A scheme displaying the possible topologies for the two short fragments TM1 and TM12 of the Y4 receptor, and experimental data on the location of the C-terminus are depicted in Fig. 6B and D.
In case of TM1, two topologies are possible for each fusion (Fig. 6B): a dual topology is observed in case of fusion with Lep (Fig. 6B and C), while a predominant cytosolic location exists for Mistic-TM1 (Fig. 6D and E). Conversely, for TM12 when fused to Lep, it predominantly adopts a periplasmic location (Fig. 6B and C), while when fused to Mistic it adopts dual topology (Fig. 6D). Possible orientation of the TM helices with respect to the E. coli inner membrane, considering the positive inside rule91 are reported (Fig. 6B and D). Our topology data are in good agreement with predictions of insertion based on the free energies of transferring entire TM helices into the membrane (Fig. 6F).92,93 They reveal that the first three helices, and in particular TM1, are unlikely to insert, while the sequences of TM4–6 strongly favor insertion.
Discussion
During membrane protein folding, interactions both with water as well as with lipids are relevant. Membrane protein biosynthesis is a vectorial process, and hence not all native (polar) tertiary contacts may be possible when the first TM helices are ready for insertion into the membrane. Therefore, non-native interactions with water or lipids may be formed that could possibly influence the folding pathway.In eukaryotes membrane protein biosynthesis occurs at the ribosome in the ER starting at the N terminus from which the nascent chain is transferred via the signal recognition particle (SRP), which targets the nascent chain to the ER inner membrane.94,95 From the SRP the chain is transferred to the large channel of the translocon machinery (Fig. 7A). In the original co-translational folding model, the emerging polypeptide chain is translocated or partitioned into the hydrophobic environment of the membrane bilayer, where the translocon decides which parts of the chain to translocate and which to insert.96–98 The insertion of helices into hydrophobic environments depends on biophysical properties of the amino acid sequence in the corresponding stretch. Wimley and White experimentally determined the free energy of transferring amino acids into the membrane interior or the membrane interface.92,93 Similarly, von Heijne has established an in vivo translocon-based scale, that qualitatively largely agrees with the in vitro data.99,100 Both scales qualitatively describe the membrane integration behavior and conformational preferences surprisingly well.
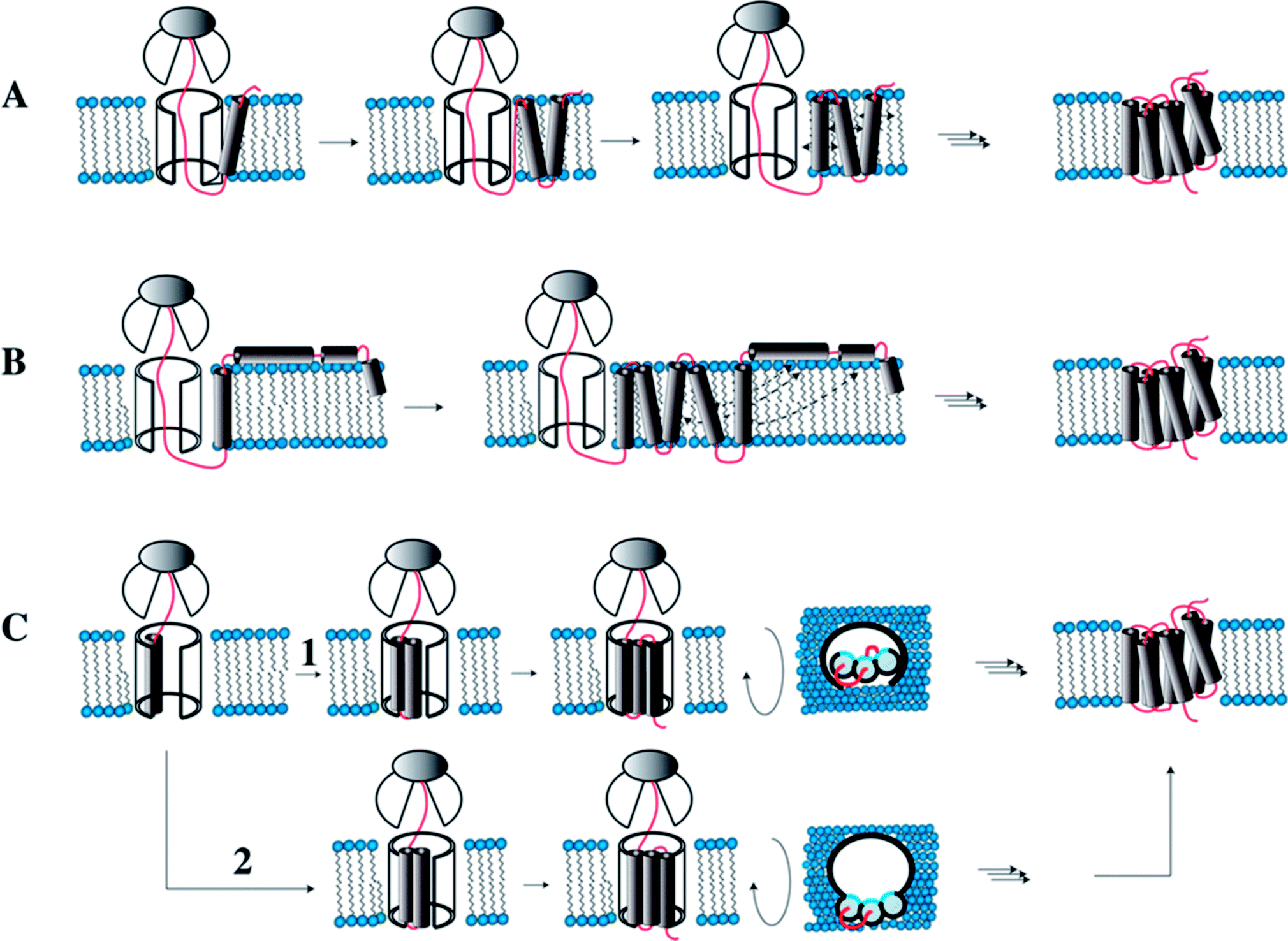 | ||
| Fig. 7 Models for co-translational folding. (A) TM helix segments are immediately released into the hydrophobic core, remain there and diffuse in the membrane until proper inter-helical contacts are made. (B) Immediate release of TM segments into the hydrophobic core. Depending on hydrophobicity some will locate in the interface. Once all TMs are made and helices transiently located in the interface, they will fully insert when proper contacts are made. (C) TM helices remain associated within the translocon until all helices are made, and then the bundle is transferred into the hydrophobic core. In (1) the TMs accumulate inside the translocon, while in (2) they are stored outside the translocon such that the hydrophilic sides (shown in blue) point into the interior of the channel. Figure modified from Skach.94 | ||
Engelman and Popot in their two-stage model for folding of helical membrane proteins41,101 postulated that helices spontaneously form once the segments coding for the TM helices insert into the membrane. This is the case because formation of secondary structure prevents that polar moieties of the peptide bond form unfavorable contacts with lipids. Following the coupled insertion/folding step helices diffuse in the membrane until the proper interhelical contacts are formed. In this simplest form the model requires that helices remain inserted until the entire chain has been biosynthesized, all inter-helical contacts are formed, and folding is completed. Recent evidence, however, indicates that individual helices of integral membrane proteins are often marginally stable in the hydrophobic interior,102 and many helices are not even expected to insert at all. The reason for this at first sight surprising property is the fact that TM helices often contain polar or even charged residues – these mediate the specific inter-helical contacts that drive the assembly of the helical bundle.103–106 It is a legitimate question to ask what happens with these marginally stable TM helices when other helices that provide the compensating interactions are not present because they have not been synthesized yet. Will they remain in the hydrophobic interior, or will they become surface associated? If they partition into the interface, will they fully re-insert once the missing TMs have been synthesized? Or do they need to be stored close to the translocon until all parts of the chain have become available?
We propose that these questions can be best addressed when studying protein fragments. These offer the conceptual advantage, when compared to the study of the entire proteins under slightly denaturing conditions, that parts, which are biosynthesized later, are not contained in the construct and hence cannot form any interactions. Solution NMR studies of fragments in membrane-mimicking environments allow studying structural details of early folding intermediates. In this review we have outlined our efforts to study folding and recognition of GPCRs from fragments.
To summarize, we have learnt that secondary structure in these fragments largely corresponds to the prediction from homology models. Hence, secondary structure is likely encoded in the local sequence, and does not completely depend on the formation of tertiary contacts as in the case of soluble proteins. In contrast, tertiary contacts are mostly not formed, and the occurrence and persistence of these contacts is related to the location and number of polar or charged residues located within the hydrophobic compartment of the membrane. The work on TM1, TM12 and TM123 or TM127 of Ste2p indicates that longer fragments integrate better into the membrane and more likely form tertiary structure. Similarly, the systematic topogenesis study of the series of Y4 receptor fragments clearly indicates that fragments containing more TM helices insert more stably and in a more defined topology. Importantly, TM1 of the Y4 receptor is predicted to not partition into the membrane,107 and when taking this feature into account the topology of the truncated fragments can be predicted rather reliably (see Fig. 7).
Inside the translocon, hydrophobic segments are laterally gated through a cleft into the membrane interior.108 Which segments are partitioned into the membrane interior is believed to be primarily decided based on hydrophobicity, a property that is reflected in the biophysical properties of the amino acid sequence.109 If TM helices are sufficiently hydrophobic they will remain inserted in the membrane and diffuse until the native contacts are made and the protein has assumed its final topology (Fig. 7A). However, when TM helices contain charged or many polar residues they may not properly insert and rather become surface associated. Once all remaining TM helices are released by the translocon, these surface-associated helices may form the compensating interactions and re-insert (topological maturation94) (Fig. 7B). Alternatively, TM segments may remain within or near to the translocon (Fig. 7C) to be released into the hydrophobic interior only once the entire polypeptide chain has been made (Fig. 7C).94
Interesting experimental results concerning cellular mechanisms of α-helical membrane protein biogenesis were obtained on the two closely related water-channel proteins aquaporin-1 (AQP1) and aquaporin-4 (AQP4). Using truncated fragments it was demonstrated that TM segments of AQP4 leave the translocon one after another in a perfectly sequential mode, and insert into the membrane in the correct topology (corresponding to Fig. 7A).110,111 In contrast, TM2 of AQP1 does not terminate translocation and therefore is partitioned into the ER lumen instead of into the membrane resulting in a four-TM instead of the native 6-TM bundle.112 The group of Dowhan has demonstrated how changes in the composition of the phospholipids switch the topology of helical membrane proteins in a non-assisted manner,113,114 and hence spontaneous topology maturation may occur (Fig. 7B). On the other hand, photo-crosslinking experiments indicated that TM helices might form either specific or nonspecific contacts with residues from the translocon,115 and that TM helix bundles could leave the translocon and enter the lipid bilayer in a concerted manner.110,116–118 Using chemical cross-linking it was also shown that large portions of opsin remain bound or associated to the translocon during ribosomal synthesis.119 Recent data on the structure of a ribosome-nascent chain translocon complex revealed that TM1 and TM2 of proteorhodopsin inserted into the membrane close to the lateral gate.120 Based on this observation von Heijne and White have suggested that TM helices never fully insert into the translocon but rather slide along the lateral gate.121 Possibly, polar side chains may then still point into the translocon channel in order to escape forming contacts to lipids, while most of the hydrophobic part would be placed rather outside the translocon (Fig. 7C (path 2)). We noticed that such a scenario would help to reduce space requirements for all TMs in the translocon interior. Our data will not allow deciding on the exact details of Ste2p or Y4 GPCR biogenesis, but they provide molecular details of early states during the folding in a valid environment and help understanding the underlying biophysics.
In principle, recognition of peptide ligands by their GPCRs again concerns the interaction of polypeptide chains in a lipid environment, this time of course for separate chains. Our studies revealed interactions of the pancreatic polypeptide PP with the N-terminal extracellular domain. These suggest that the ligand is transferred via a fly-casting mechanism from the membrane-associated state into the ligand-binding pocket, which is mostly formed by the extracellular loops. In fragments the orthosteric ligand-binding site is either not present or not fully formed, and hence this weak interaction, while likely short-lived in the entire receptor, can still be observed. The grafting work demonstrates that extracellular loops provide binding affinity for the ligands when provided in an approximately correct topology. However, the work also showed that the binding affinity for the grafted model is significantly lower than for the entire receptor. This work therefore highlights a constant dilemma when working with fragments – they provide much conceptual insight into folding or binding but can never fully reproduce the natural system. Nevertheless, we believe that the information gained on these systems justifies using fragments to address specific questions of folding and binding.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The described work on the Ste2p receptor was performed in very fruitful collaboration with the laboratories of Fred Naider and Jeff Becker. I deeply acknowledge their crucial contributions to the projects. The described work has been supported by the Swiss National Science Foundation (grants No. 31003A_124649 and 310030_159453 to O.Z.).References
- K. Sriram and P. A. Insel, Mol. Pharmacol., 2018, 93, 251–258 CrossRef PubMed.
- A. J. Venkatakrishnan, X. Deupi, G. Lebon, F. M. Heydenreich, T. Flock, T. Miljus, S. Balaji, M. Bouvier, D. B. Veprintsev, C. G. Tate, G. F. Schertler and M. M. Babu, Nature, 2016, 536, 484–487 CrossRef CAS PubMed.
- T. Flock, A. S. Hauser, N. Lund, D. E. Gloriam, S. Balaji and M. M. Babu, Nature, 2017, 545, 317–322 CrossRef CAS PubMed.
- G. F. Schertler, C. Villa and R. Henderson, Nature, 1993, 362, 770–772 CrossRef CAS PubMed.
- K. Palczewski, T. Kumasaka, T. Hori, C. A. Behnke, H. Motoshima, B. A. Fox, I. Le Trong, D. C. Teller, T. Okada, R. E. Stenkamp, M. Yamamoto and M. Miyano, Science, 2000, 289, 739–745 CrossRef CAS PubMed.
- B. Kobilka, Angew. Chem., Int. Ed. Engl., 2013, 52, 6380–6388 CrossRef CAS PubMed.
- R. J. Lefkowitz, Angew. Chem., Int. Ed. Engl., 2013, 52, 6366–6378 CrossRef CAS PubMed.
- R. Grisshammer, Protein Sci., 2017, 26, 1493–1504 CrossRef CAS PubMed.
- K. S. Huang, H. Bayley, M. J. Liao, E. London and H. G. Khorana, J. Biol. Chem., 1981, 256, 3802–3809 CAS.
- O. Tastan, A. Dutta, P. Booth and J. Klein-Seetharaman, Biochim. Biophys. Acta, Bioenerg., 2014, 1837, 656–663 CrossRef CAS PubMed.
- N. R. Latorraca, A. J. Venkatakrishnan and R. O. Dror, Chem. Rev., 2017, 117, 139–155 CrossRef CAS PubMed.
- A. L. Klyszejko, S. Shastri, S. A. Mari, H. Grubmüller, D. J. Müller and C. Glaubitz, J. Mol. Biol., 2007, 376, 35–41 CrossRef PubMed.
- D. Fotiadis, B. Jastrzebska, A. Philippsen, D. J. Müller, K. Palczewski and A. Engel, Curr. Opin. Struct. Biol., 2006, 16, 252–259 CrossRef CAS PubMed.
- D. J. Müller, K. T. Sapra, S. Scheuring, A. Kedrov, P. L. Frederix, D. Fotiadis and A. Engel, Curr. Opin. Struct. Biol., 2006, 16, 489–495 CrossRef PubMed.
- O. Zerbe, Angew. Chem., Int. Ed. Engl., 2011, 51, 860–861 CrossRef PubMed.
- A. Gautier, H. R. Mott, M. J. Bostock, J. P. Kirkpatrick and D. Nietlispach, Nat. Struct. Mol. Biol., 2010, 17, 768–774 CAS.
- S. Reckel, D. Gottstein, J. Stehle, F. Löhr, M. K. Verhoefen, M. Takeda, R. Silvers, M. Kainosho, C. Glaubitz, J. Wachtveitl, F. Bernhard, H. Schwalbe, P. Güntert and V. Dötsch, Angew. Chem., Int. Ed. Engl., 2011, 50, 11942–11946 CrossRef CAS PubMed.
- R. Nygaard, Y. Zou, R. O. Dror, T. J. Mildorf, D. H. Arlow, A. Manglik, A. C. Pan, C. W. Liu, J. J. Fung, M. P. Bokoch, F. S. Thian, T. S. Kobilka, D. E. Shaw, L. Mueller, R. S. Prosser and B. K. Kobilka, Cell, 2013, 152, 532–542 CrossRef CAS PubMed.
- R. Sounier, C. Mas, J. Steyaert, T. Laeremans, A. Manglik, W. Huang, B. K. Kobilka, H. Déméné and S. Granier, Nature, 2015, 524, 375–378 CrossRef CAS PubMed.
- M. P. Bokoch, Y. Zou, S. G. F. Rasmussen, C. W. Liu, R. Nygaard, D. M. Rosenbaum, J. J. Fung, H.-J. Choi, F. S. Thian, T. S. Kobilka, J. D. Puglisi, W. I. Weis, L. Pardo, R. S. Prosser, L. Mueller and B. K. Kobilka, Nature, 2010, 463, 108–112 CrossRef CAS PubMed.
- S. Isogai, X. Deupi, C. Opitz, F. M. Heydenreich, C. J. Tsai, F. Brueckner, G. F. Schertler, D. B. Veprintsev and S. Grzesiek, Nature, 2016, 530, 237–241 CrossRef CAS PubMed.
- M. Casiraghi, M. Damian, E. Lescop, E. Point, K. Moncoq, N. Morellet, D. Levy, J. Marie, E. Guittet, J. L. Banères and L. J. Catoire, J. Am. Chem. Soc., 2016, 138, 11170–11175 CrossRef CAS PubMed.
- J. J. Liu, R. Horst, V. Katritch, R. C. Stevens and K. Wüthrich, Science, 2012, 335, 1106–1110 CrossRef CAS PubMed.
- L. Ye, N. Van Eps, M. Zimmer, O. P. Ernst and R. S. Prosser, Nature, 2016, 533, 265–268 CrossRef CAS PubMed.
- M. D. Turton, D. O'Shea and S. R. Bloom, Neuropept. Y Drug Dev., 1997, 15–39 CAS.
- R. Bader and O. Zerbe, ChemBioChem, 2005, 6, 1520–1534 CrossRef CAS PubMed.
- D. F. Sargent and R. Schwyzer, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 5774–5778 CrossRef CAS.
- R. Schwyzer, J. Mol. Recognit., 1995, 8, 3–8 CrossRef CAS PubMed.
- F. Naider and J. M. Becker, Peptides, 2004, 25, 1441–1463 CrossRef CAS PubMed.
- R. Koebnik, J. Bacteriol., 1999, 181, 3688–3694 CAS.
- M. U. Johansson, S. Alioth, K. Hu, R. Walser, R. Koebnik and K. Pervushin, Biochemistry, 2007, 46, 1128–1140 CrossRef CAS PubMed.
- R. Walser, J. H. Kleinschmidt and O. Zerbe, ChemBioChem, 2011, 12, 1690–1693 CrossRef CAS PubMed.
- R. Walser, J. H. Kleinschmidt, A. Skerra and O. Zerbe, Biol. Chem., 2012, 393, 1341–1355 CrossRef CAS PubMed.
- R. Sprangers and L. E. Kay, Nature, 2007, 445, 618–622 CrossRef CAS PubMed.
- S. H. White and W. C. Wimley, Annu. Rev. Biophys. Biomol. Struct., 1999, 28, 319–365 CrossRef CAS PubMed.
- J. L. Popot and D. M. Engelman, Annu. Rev. Biochem., 2000, 69, 881–922 CrossRef CAS PubMed.
- T. Marti, J. Biol. Chem., 1998, 273, 9312–9322 CrossRef CAS PubMed.
- N. P. Martin, L. M. Leavitt, C. M. Sommers and M. E. Dumont, Biochemistry, 1999, 38, 682–695 CrossRef CAS PubMed.
- P. L. Yeagle, J. L. Alderfer and A. D. Albert, Nat. Struct. Biol., 1995, 2, 832–834 CrossRef CAS PubMed.
- B. König, A. Arendt, J. H. McDowell, M. Kahlert, P. A. Hargrave and K. P. Hofmann, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 6878–6882 CrossRef.
- J. L. Popot and D. M. Engelman, Biochemistry, 1990, 29, 4031–4037 CrossRef CAS PubMed.
- K. R. MacKenzie, Chem. Rev., 2006, 106, 1931–1977 CrossRef CAS PubMed.
- K. R. Mackenzie, J. H. Prestegard and D. M. Engelman, Science, 1997, 276, 131–133 CrossRef CAS PubMed.
- D. Langosch and I. T. Arkin, Protein Sci., 2009, 18, 1343–1358 CrossRef CAS PubMed.
- M. A. Lemmon, J. M. Flanagan, H. R. Treutlein, J. Zhang and D. M. Engelman, Biochemistry, 1992, 31, 12719–12725 CrossRef CAS PubMed.
- W. P. Russ and D. M. Engelman, J. Mol. Biol., 2000, 296, 911–919 CrossRef CAS PubMed.
- D. Langosch and J. Heringa, Proteins, 1998, 31, 150–159 CrossRef CAS.
- D. A. Dougherty, Science, 1996, 271, 163–168 CAS.
- J. Bowie, Nature, 2005, 438, 581–589 CrossRef CAS PubMed.
- X. Shao, C. Zou, F. Naider and O. Zerbe, Biophys. J., 2012, 103, 817–826 CrossRef CAS PubMed.
- H. Kocherla, J. Marino, X. Shao, J. Graf, C. Zou and O. Zerbe, ChemBioChem, 2012, 13, 818–828 CrossRef CAS PubMed.
- L. Cohen, K. Fracchiolla, J. Becker and F. Naider, Biopolymers, 2014, 102, 223–243 CrossRef CAS PubMed.
- J. Zhao, H. Zheng and X.-Q. Xie, Protein Pept. Lett., 2006, 13, 335–342 CrossRef CAS PubMed.
- Y. Zhang and X.-Q. Xie, Protein Expression Purif., 2008, 59, 249–257 CrossRef CAS PubMed.
- H. Xie, F. X. Ding, D. Schreiber, G. Eng, S. F. Liu, B. Arshava, E. Arevalo, J. M. Becker and F. Naider, Biochemistry, 2000, 39, 15462–15474 CrossRef CAS PubMed.
- A. Kerman and V. S. Ananthanarayanan, Biochim. Biophys. Acta, 2005, 1747, 133–140 CrossRef CAS PubMed.
- A. Kerman and V. S. Ananthanarayanan, Biochim. Biophys. Acta, 2007, 1768, 1199–1210 CrossRef CAS PubMed.
- G. F. Miozzari and C. Yanofsky, J. Bacteriol., 1978, 133, 1457–1466 CAS.
- T. Roosild, J. Greenwald, M. Vega, S. Castronovo, R. Riek and S. Choe, Science, 2005, 307, 1317–1321 CrossRef CAS PubMed.
- R. Estephan, J. Englander, B. Arshava, K. L. Samples, J. M. Becker and F. Naider, Biochemistry, 2005, 44, 11795–11810 CrossRef CAS PubMed.
- H. Kiefer, Biochim. Biophys. Acta, 2003, 1610, 57–62 CrossRef CAS.
- P. J. Booth, Biochim. Biophys. Acta, 2003, 1610, 51–56 CrossRef CAS.
- R. Grisshammer, Methods Enzymol., 2009, 463, 631–645 CAS.
- R. Bader, A. Bettio, A. G. Beck-Sickinger and O. Zerbe, J. Mol. Biol., 2001, 305, 307–392 CrossRef CAS PubMed.
- M. Lerch, V. Gafner, R. Bader, B. Christen, G. Folkers and O. Zerbe, J. Mol. Biol., 2002, 322, 1117–1133 CrossRef CAS PubMed.
- M. Lerch, M. Mayrhofer and O. Zerbe, J. Mol. Biol., 2004, 339, 1153–1168 CrossRef CAS PubMed.
- A. Neumoin, J. Mares, M. Lerch-Bader, R. Bader and O. Zerbe, J. Am. Chem. Soc., 2007, 129, 8811–8817 CrossRef CAS PubMed.
- L. Moroder, R. Romano, W. Guba, D. Mierke, H. Kessler, C. Delporte, J. Winand and J. Christophe, Biochemistry, 1993, 32, 13551–13559 CrossRef CAS PubMed.
- R. Kage, S. E. Leeman, J. E. Krause, C. E. Costello and N. D. Boyd, J. Biol. Chem., 1996, 271, 25797–25800 CrossRef CAS PubMed.
- H. Li, D. M. Macdonald, X. Hronowski, C. E. Costello, S. E. Leeman and N. D. Boyd, J. Biol. Chem., 2001, 276, 10589–10593 CrossRef CAS PubMed.
- D. Macdonald, D. F. Mierke, H. Li, M. Pellegrini, B. Sachais, J. E. Krause, S. E. Leeman and N. D. Boyd, Biochemistry, 2001, 40, 2530–2539 CrossRef CAS PubMed.
- C. Zou, S. Kumaran, R. Walser and O. Zerbe, J. Pept. Sci., 2009, 15, 184–191 CrossRef CAS PubMed.
- C. Zou, S. Kumaran, S. Markovic, R. Walser and O. Zerbe, ChemBioChem, 2008, 9, 2276–2284 CrossRef CAS PubMed.
- S. Boivin, L. Guilhaudis, I. Milazzo, H. Oulyadi, D. Davoust and A. Fournier, Biochemistry, 2006, 45, 5993–6002 CrossRef CAS PubMed.
- B. Wu, E. Y. Chien, C. D. Mol, G. Fenalti, W. Liu, V. Katritch, R. Abagyan, A. Brooun, P. Wells, F. C. Bi, D. J. Hamel, P. Kuhn, T. M. Handel, V. Cherezov and R. C. Stevens, Science, 2010, 330, 1066–1071 CrossRef CAS PubMed.
- N. A. Diepenhorst, P. R. Gooley, M. J. Stone and R. A. Bathgate, Ital. J. Anat. Embryol., 2013, 118, 1–3 Search PubMed.
- N. A. Diepenhorst, E. J. Petrie, C. Z. Chen, A. Wang, M. A. Hossain, R. A. Bathgate and P. R. Gooley, J. Biol. Chem., 2014, 289, 34938–34952 CrossRef PubMed.
- C. O. Audu, J. J. Plati, M. Pellegrini and D. F. Mierke, Proteins, 2014, 82, 1370–1375 CrossRef CAS PubMed.
- A. Groß, R. Brox, D. Damm, N. Tschammer, B. Schmidt and J. Eichler, Bioorg. Med. Chem., 2015, 23, 4050–4055 CrossRef PubMed.
- K. Möbius, R. Dürr, C. Haussner, U. Dietrich and J. Eichler, Chem.–Eur. J., 2012, 18, 8292–8295 CrossRef PubMed.
- M. Poms, P. Ansorge, L. Martinez-Gil, S. Jurt, D. Gottstein, K. E. Fracchiolla, L. S. Cohen, P. Güntert, I. Mingarro, F. Naider and O. Zerbe, J. Biol. Chem., 2016, 291, 27170–27186 CrossRef CAS PubMed.
- M. Eilers, V. Hornak, S. O. Smith and J. B. Konopka, Biochemistry, 2005, 44, 8959–8975 CrossRef CAS PubMed.
- A. Neumoin, B. Arshava, J. Becker, O. Zerbe and F. Naider, Biophys. J., 2007, 93, 467–482 CrossRef CAS PubMed.
- C. Zou, F. Naider and O. Zerbe, J. Biomol. NMR, 2008, 42, 257–269 CrossRef CAS PubMed.
- M. J. Liao, K. S. Huang and H. G. Khorana, J. Biol. Chem., 1984, 259, 4200–4204 CAS.
- A. Neumoin, L. S. Cohen, B. Arshava, S. Tantry, J. M. Becker, O. Zerbe and F. Naider, Biophys. J., 2009, 96, 3187–3196 CrossRef CAS PubMed.
- Z. Potetinova, S. Tantry, L. S. Cohen, K. E. Caroccia, B. Arshava, J. M. Becker and F. Naider, Biopolymers, 2012, 98, 485–500 CrossRef CAS PubMed.
- D. Drew, D. Sjostrand, J. Nilsson, T. Urbig, C. N. Chin, J. W. D. Gier and G. von Heijne, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 2690–2695 CrossRef CAS PubMed.
- D. E. Drew, G. von Heijne, P. Nordlund and J.-W. L. de Gier, FEBS Lett., 2001, 507, 220–224 CrossRef CAS PubMed.
- N. Bilgin, J. I. Lee, H. Y. Zhu, R. Dalbey and G. von Heijne, EMBO J., 1990, 9, 2717–2722 CAS.
- G. von Heijne, Nature, 1989, 341, 456–458 CrossRef CAS PubMed.
- S. H. White and W. C. Wimley, Biochim. Biophys. Acta, 1998, 1376, 339–352 CrossRef CAS.
- W. C. Wimley and S. H. White, Nat. Struct. Biol., 1996, 3, 842–848 CrossRef CAS PubMed.
- W. R. Skach, Nat. Struct. Mol. Biol., 2009, 16, 606–612 CAS.
- G. von Heijne, Q. Rev. Biophys., 1999, 32, 285–307 CrossRef CAS PubMed.
- B. Van den Berg, W. M. Clemons, I. Collinson, Y. Modis, E. Hartmann, S. C. Harrison and T. A. Rapoport, Nature, 2004, 427, 36–44 CrossRef CAS PubMed.
- T. A. Rapoport, V. Goder, S. U. Heinrich and K. E. Matlack, Trends Cell Biol., 2004, 14, 568–575 CrossRef CAS PubMed.
- P. F. Egea and R. M. Stroud, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17182–17187 CrossRef CAS PubMed.
- T. Hessa, H. Kim, K. Bihlmaier, C. Lundin, J. Boekel, H. Andersson, I. Nilsson, S. H. White and G. von Heijne, Nature, 2005, 433, 377–381 CrossRef CAS PubMed.
- T. Hessa, N. M. Meindl-Beinker, A. Bernsel, H. Kim, Y. Sato, M. Lerch-Bader, I. Nilsson, S. H. White and G. von Heijne, Nature, 2007, 450, 1026–1030 CrossRef CAS PubMed.
- D. M. Engelman, Y. Chen, C. N. Chin, A. R. Curran, A. M. Dixon, A. D. Dupuy, A. S. Lee, U. Lehnert, E. E. Matthews, Y. K. Reshetnyak, A. Senes and J. L. Popot, FEBS Lett., 2003, 555, 122–125 CrossRef CAS PubMed.
- L. E. Hedin, K. Ojemalm, A. Bernsel, A. Hennerdal, K. Illergård, K. Enquist, A. Kauko, S. Cristobal, G. von Heijne, M. Lerch-Bader, I. Nilsson and A. Elofsson, J. Mol. Biol., 2010, 396, 221–229 CrossRef CAS PubMed.
- M. Bano-Polo, L. Martinez-Gil, B. Wallner, J. L. Nieva, A. Elofsson and I. Mingarro, J. Mol. Biol., 2013, 425, 830–840 CrossRef CAS PubMed.
- A. Senes, D. E. Engel and W. F. DeGrado, Curr. Opin. Struct. Biol., 2004, 14, 465–479 CrossRef CAS PubMed.
- F. X. Zhou, H. J. Merianos, A. T. Brünger and D. M. Engelman, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 2250–2255 CrossRef CAS PubMed.
- V. Krishnamani and J. K. Lanyi, Biochemistry, 2012, 51, 1061–1069 CrossRef CAS PubMed.
- J. Marino, E. R. Geertsma and O. Zerbe, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 3055–3063 CrossRef CAS PubMed.
- A. R. Osborne, T. A. Rapoport and B. van den Berg, Annu. Rev. Cell Dev. Biol., 2005, 21, 529–550 CrossRef CAS PubMed.
- S. H. White and G. von Heijne, Annu. Rev. Biophys., 2008, 37, 23–42 CrossRef CAS PubMed.
- H. Sadlish, D. Pitonzo, A. E. Johnson and W. R. Skach, Nat. Struct. Mol. Biol., 2005, 12, 870–878 CAS.
- L. B. Shi, W. R. Skach, T. Ma and A. S. Verkman, Biochemistry, 1995, 34, 8250–8256 CrossRef CAS PubMed.
- W. R. Skach, L. B. Shi, M. C. Calayag, A. Frigeri, V. R. Lingappa and A. S. Verkman, J. Cell Biol., 1994, 125, 803–815 CrossRef CAS PubMed.
- M. Bogdanov, J. Xie, P. Heacock and W. Dowhan, J. Cell Biol., 2008, 182, 925–935 CrossRef CAS PubMed.
- H. Vitrac, D. M. MacLean, V. Jayaraman, M. Bogdanov and W. Dowhan, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 13874–13879 CrossRef CAS PubMed.
- P. J. McCormick, Y. Miao, Y. Shao, J. Lin and A. E. Johnson, Mol. Cell, 2003, 12, 329–341 CrossRef CAS PubMed.
- D. Pitonzo, Z. Yang, Y. Matsumura, A. E. Johnson and W. R. Skach, Mol. Biol. Cell, 2009, 20, 685–698 CrossRef CAS PubMed.
- A. Sauri, S. Saksena, J. Salgado, A. E. Johnson and I. Mingarro, J. Biol. Chem., 2005, 280, 25907–25912 CrossRef CAS PubMed.
- A. Sauri, P. J. McCormick, A. E. Johnson and I. Mingarro, J. Mol. Biol., 2007, 366, 366–374 CrossRef CAS PubMed.
- N. Ismail, S. G. Crawshaw, B. C. Cross, A. C. Haagsma and S. High, Biochem. J., 2008, 411, 495–506 CrossRef CAS PubMed.
- L. Bischoff, S. Wickles, O. Berninghausen, E. O. van der Sluis and R. Beckmann, Nat. Commun., 2014, 5, 4103 CAS.
- F. Cymer, G. von Heijne and S. H. White, J. Mol. Biol., 2015, 427, 999–1022 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |