
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn efficient and scalable synthesis of potent TLR2 agonistic PAM2CSK4†
Arshpreet Kaura,
Poonama,
Madhuri T. Patilb,
Surinder K. Mehta a and
Deepak B. Salunke*a
a and
Deepak B. Salunke*a
aDepartment of Chemistry and Centre of Advanced Studies in Chemistry, Panjab University, Chandigarh 160014, India. E-mail: salunke@pu.ac.in
bDepartment of Chemistry, Mehr Chand Mahajan DAV College for Women, Sector 36A, Chandigarh 160036, India
First published on 5th March 2018
Abstract
Diacylated PAM2CSK4, a highly expensive lipopeptide with desirable aqueous solubility and a broad spectrum of cytokine/chemokine induction is a most potent dual (human and murine) Toll-Like Receptor-2 (TLR2) agonist. Besides such thrilling characteristics, its synthetic process is not reported in the literature. The present report describes an efficient and scalable 20 step synthesis of PAM2CSK4 in good yield (all steps > 60%) along with a clear description of the hindrances and easy solutions adopted in each step. Overall, a convergent synthetic approach was adopted involving synthesis of appropriately protected starting materials, synthesis of a key backbone skeleton PAM2CS, synthesis of a tetralysine fragment and the final coupling to yield PAM2CSK4. Tedious column chromatography was avoided on a large scale in many steps.
Introduction
The current scenario of a war zone against pathogenic infections demands the enactment of an innate and adaptive immune system and their working as a two-wheel drive. As one of the key players of the innate immune system, Toll-Like Receptors (TLRs) present in the immune cells perceive pathogen associated molecular patterns (PAMPs) conserved in microbes.1 The recognition of PAMPs through these Pattern Recognition Receptors (PRRs) brings out the induction of a signaling cascade with the production of cytokines and costimulatory molecules.2 Thereafter, the activation of an adaptive immune system occurs through TLRs, which serve as a bridge between two immune systems. TLRs are the membrane spanning proteins situated on the cell surface of immune cells or in endosomal compartments. Each TLR is amendable to perceive a specific set of pathogens with the variation in inflammatory responses.3 An overwhelming inflammatory response is expected from the upcoming novel immunotherapeutics or vaccine adjuvants.4 Therefore, the principle emphasis of numerous specialists in the field of medicinal chemistry is the design and synthesis of ligands that specifically activate TLRs.Due to the complexity related to mechanistic approaches to combat any particular disease, the choice of appropriate target from total 13 known TLRs (11 in humans and 13 in mice) is a major concern for immunologists. Natural ligands for all the TLRs have been identified except for TLR10. These ligands include lipopeptides (recognized by TLR2 in combination with TLR1 or TLR6), lipopolysaccharides (LPS) (TLR4), microbial protein components such as flagellin (TLR5), nucleic acids, for example, single-stranded RNA (TLR7 and TLR8) or double-stranded RNA (TLR3) and CpG DNA (TLR9).5
Among all the human TLRs, TLR2 is known to sense remarkable array of bacterial, fungal, and viral products as well as inflammatory self-segments.6 TLR2 is receptor equipped for experiencing heterodimerization with TLR1 (for triacylated lipopeptides) and TLR6 (for diacylated lipopeptides).7 Additionally, TLR2 has been widely explored as a promising target for drug discovery (Table 1). SPM-105, a TLR2 agonistic autoclaved mycobacteria was considered in preclinical examinations for the treatment of cancer8 and OM-174, a lipid A derivative with double activity on TLR2 and TLR4, was tried as a chemotherapeutic agent for viability against unmanageable strong tumors.9 Likewise, bacterial porins showing TLR2 agonistic activity have been utilized in vaccines. Specifically, OmpS1 and OmpS2, the external film proteins from Salmonella typhi, are known to actuate TLR2 and TLR4 and are used for immunization in typhoid fever.10 Among the several TLR2 agonistic biomacromolecules, the lipopeptides derived from cell-wall components are the most intense ligands.6 PAM3CSK4 (N-palmitoyl-S-[2,3-bis(palmitoyloxy)-(2RS)-propyl]-R-cysteinyl-S-seryl-S-lysyl-S-lysyl-S-lysyl-S-lysine) as well as PAM2CSK4 (S-[2,3-bis(palmitoyloxy)-(2RS)-propyl]-R-cysteinyl-S-seryl-S-lysyl-S-lysyl-S-lysyl-S-lysine) are the greatly explored synthetic lipopeptides used against parasitic infections such as leishmania,11 malaria,12 influenza13 and also worked well in suppression of asthma manifestations14 and melanoma15 (Table 1).
| Compound | Target | Indication | Drug class |
|---|---|---|---|
| SMP-105 | TLR2 agonist | Bladder cancer | Autoclaved mycobacteria8 |
| OM-174 | TLR2 & 4 agonist | Cancer chemotherapy | Lipid A derivative9 |
| OmpS1 & OmpS2 (Salmonella typhi) | TLR2 & 4 agonist | Typhoid fever vaccine | Bacterial porins10 |
| PAM3CSK4/PAM2CSK4 | TLR2 agonist | Leishmania infection | Lipoprotein11 |
| PAM3Cys/PAM2Cys | TLR2 agonist | Malaria LbL vaccine | Lipoprotein conjugate12 |
| PAM2CSK4 | TLR2 agonist | Influenza vaccine | Lipoprotein13 |
| PAM3Cys | TLR2 agonist | Suppression of asthma manifestations | Lipoprotein14 |
| PAM3CSK4 | TLR2 agonist | Melanoma cancer | Lipoprotein15 |
Several palmitoyl lipopeptides are being explored as TLR2 agonists. Out of the analogs PAM3Cys-Ser-Ser-Asn-Ala, PAM3Cys-Ser-(Lys)4, PAM3Cys-Ala-Gly, and PAM3Cys-Ser, PAM3CSK4 was found to be a potent activator due to the presence of hydrophilic lysine residues that enhanced its solubility.16 On the other hand, the diacylated PAM2CSK4 lipopeptide has better aqueous solubility as compared to triacylated PAM3CSK4. The vaccines with peptide epitopes and dipalmitoyl lipopeptide constructs are most soluble and effective vaccine candidates.17 Moreover, dipalmitoyl lipopeptide is more potent immunostimulant than tripalmitoyl lipopeptide for macrophages and splenocytes.18 It is noteworthy to mention that an extensive SAR investigation on PAM2CSK4 resulted in highly active monoacyl lipopeptides but these molecules being human specific were not further explored in the preclinical investigation.19
After the complete review of literature, the diacylated PAM2CSK4 lipopeptide was found to be the most potent dual (human and murine) TLR2 agonist having desirable aqueous solubility showing broad spectrum of cytokine/chemokine inducing property.20 This synthetic lipopeptide is equipotent (EC50 = 67 pm)21 in its NF-κB inducing activity relative to LPS.22,23 Induction of class I cytokine IL-6, class II cytokine IL-10, IFN-β, IFN-γ, and activation of TNF receptors is reported by this ligand. PAM2CSK4 is also known to induce several chemokines namely, MCP-1, CXCL6, GRO, CXCL8, CCL20, CXCL10 and CCL5 (Table S1, page S3 ESI†).20–26 PAM2CSK4 is not only a potent TLR2 agonist but also possess property to self assemble at critical aggregation concentration (cac = 0.035 wt%). It self assemble into small spherical micelles distinct from worm-like micelles observed in case of PAM3CSK4.27 The distinct mode of self assembly may be related to its bioactivity and could be a useful parameter for the formulation.
PAM2CSK4 is highly expensive TLR2 ligand available from Invivogen28 but its synthetic process is not yet described in the literature. These observations further drove our interest to optimize the large-scale synthesis of PAM2CSK4 with minimum possible investment. The present report describes an efficient and scalable first complete synthesis of this TLR2 agonistic PAM2CSK4.
Results and discussion
PAM2CSK4, a synthetic analog of MALP-2 consists of two major components, a lipo-dipeptide PAM2CS 3 and a tetrapeptide of lysine K4 4 (Fig. 1). The retrosynthetic analysis shows that the constituents necessary for the synthesis are R-4-(iodomethyl)-2,2-dimethyl-1,3-dioxalane 5, Boc-Cys-OH 6 and H-Ser(OtBu)-OtBu 7. Based on the commercial availability and the cost of these synthons, a convergent synthetic approach involving synthesis of appropriately protected starting materials (Scheme 1), synthesis of key backbone skeleton (Scheme 2), synthesis of tetralysine fragment (Scheme 3) and final coupling to yield PAM2CSK4 (Scheme 4) was proposed for this synthesis. | ||
| Fig. 1 Retrosynthetic approach for PAM2CSK4. | ||
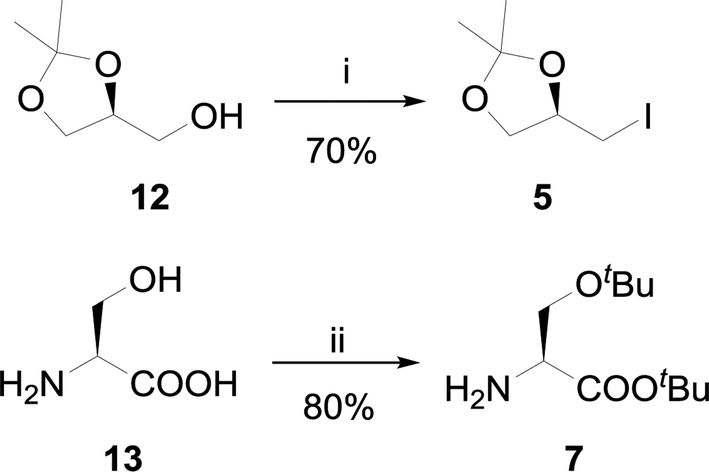 | ||
| Scheme 1 Synthesis of reagents. Conditions: (i) PPh3, I2, imidazole, THF, rt, 12 h. (ii) tert-Butyl acetate, 70% HClO4, 6 h. | ||
 | ||
| Scheme 2 Synthesis of PAM2CS. Conditions: (i) Boc2O, Et3N, H2O, rt, 12 h. (ii) H-Ser(OtBu)OtBu·HCl (7), EDCI, HOBt, Et3N, DMF, rt, 5 h. (iii) Bu3P, DCM, rt, 30 min. (iv) R-4-(Iodomethyl)-2,2-dimethyl-1,3-dioxalane (5), K2CO3, DMF, rt, 2 h. (v) 70% AcOH in H2O, rt, 12 h. (vi) C15H31COCl, Et3N, DMAP, DCM, rt, 12 h. (vii) TFA, rt, 30 min. (viii) Boc2O, Et3N, DCM, 4 h. | ||
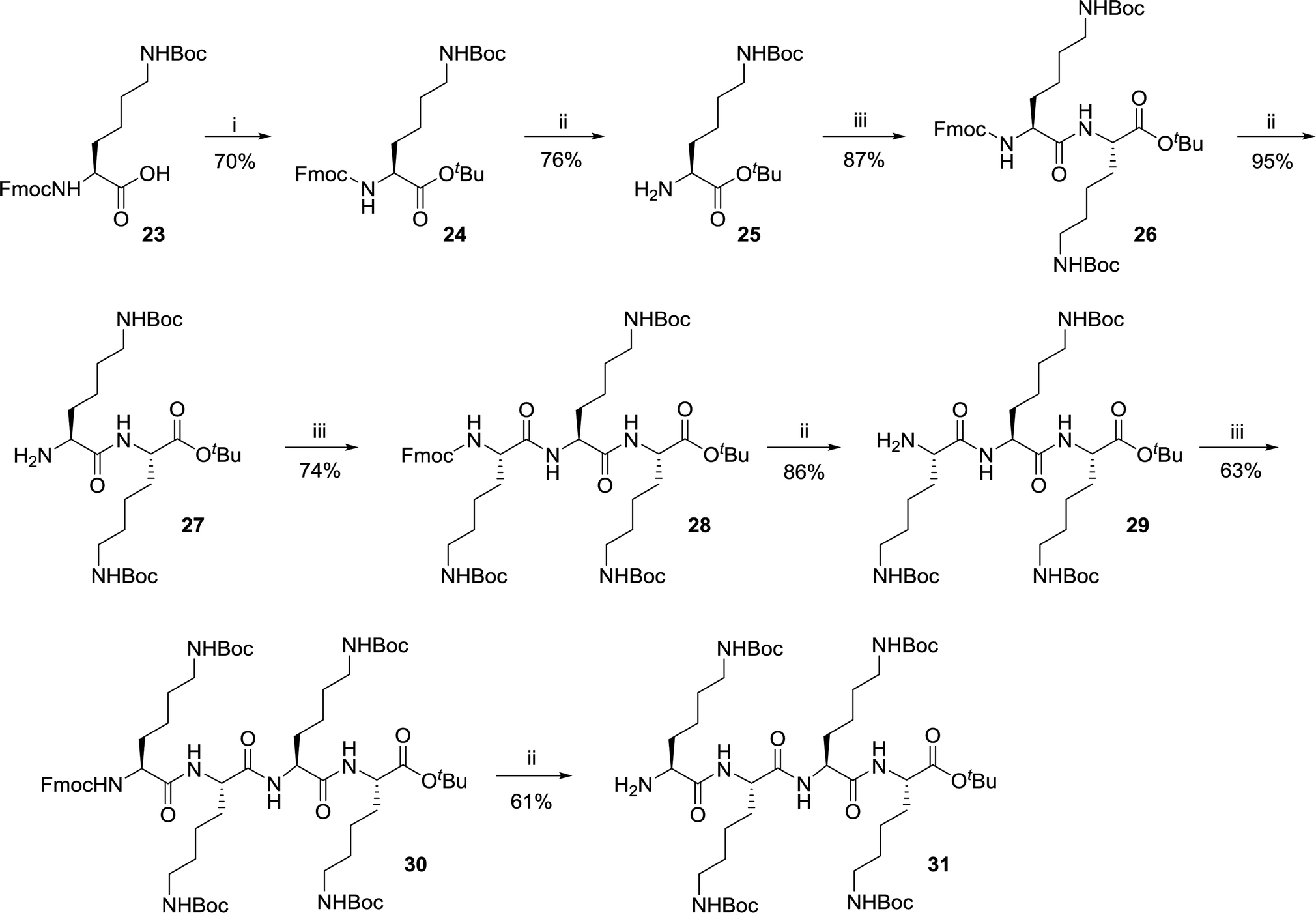 | ||
| Scheme 3 Synthesis of tertapeptide K4. Conditions: (i) Di-tert-butyl dicarbonate, DMAP, tert-butyl alcohol, rt, overnight. (ii) 20% piperidine, DMF, rt, 1 h. (iii) Fmoc-L-Lys(Boc)-OH (23), EDCI, HOBt, Et3N, DMF, rt, 6 h. | ||
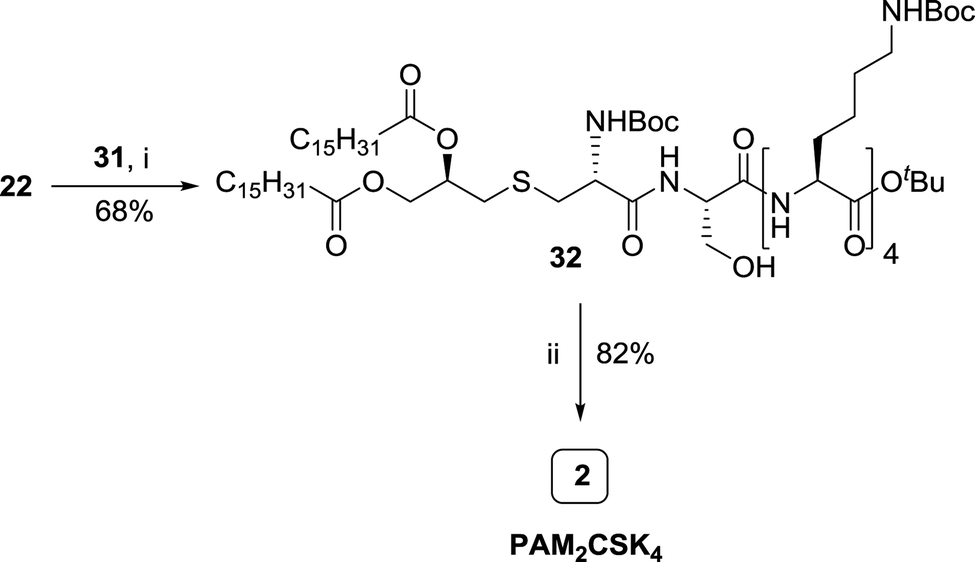 | ||
| Scheme 4 Synthesis of PAM2CSK4. Conditions: (i) H-[Lys(Boc)]4-OtBu, EDCI, HOBt, Et3N, DMF, 6 h. (ii) HCl, dioxane, 2 h. | ||
The key synthon R-4-(iodomethyl)-2,2-dimethyl-1,3-dioxalane 5 (Scheme 1) was synthesized from commercially available S-2,2-dimethyl-1,3-dioxolane-4-methanol 12 in good yield. The synthesis involved use of iodine (1.3 eq.), triphenylphosphine (1.2 eq.) and imidazole (3 eq.) reagent in toluene at 90 °C where imidazole acted as general base and an acid scavenger.29 The excess amount of iodine was necessary to enhance the formation of an alkoxyphosphonium iodide salt which slowly decomposes by SN2 mechanism to form product 5.30 A tedious column chromatography purification was also required to remove the triphenylphosphine oxide (TPPO).
The procedure was improved by replacing the solvent toluene with THF or DCM, where the reaction proceeded at room temperature and TPPO was removed by complexation using zinc chloride (ZnCl2) as described by Batesky and coworkers.31 This protocol was applied to avoid column chromatography at large scale by precipitating out triphenylphosphine oxide as ZnCl2(Ph3PO)x complex.
The high price of H-Ser(OtBu)-OtBu (5 g, Rs. 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 133 from Bachem) 7 required on large scale was another key factor to be considered in this synthesis. Several methods have been reported for the synthesis of tert-butyl ethers and esters but the distinct and easy protocol was the protection of both hydroxyl and carboxylic acid functionalities in one pot.32 The adopted procedure involved use of tert-butyl acetate for the protection of hydroxyl and carboxylic acid group of L-serine in the presence of 70% HClO4 (Scheme 1) to yield pure oily product 7 without any need of column chromatography.33
133 from Bachem) 7 required on large scale was another key factor to be considered in this synthesis. Several methods have been reported for the synthesis of tert-butyl ethers and esters but the distinct and easy protocol was the protection of both hydroxyl and carboxylic acid functionalities in one pot.32 The adopted procedure involved use of tert-butyl acetate for the protection of hydroxyl and carboxylic acid group of L-serine in the presence of 70% HClO4 (Scheme 1) to yield pure oily product 7 without any need of column chromatography.33
A lipopeptide backbone skeleton 3 mainly responsible for the agonistic activity was synthesized in 8 steps (Scheme 2). L-Cystine was used as a precursor for the synthesis wherein, the initial step involved the protection of α-amine group of L-cystine 14 (Scheme 2) using di-tert-butyl dicarbonate (Boc2O) to obtain di-Boc-cystine 15.34 After the completion of reaction, 10% HCl solution (15 mL for 2 g batch) was directly added to the reaction mixture at 0 °C and the crude product 15 was precipitated out and washed with chilled distilled water to remove impurities. No column chromatographic purification was required and the crude product 15 was used as it is for further transformations. The coupling reaction of (S)-tert-butyl-2-amino-3-tert-butoxypropanoate 7 and di-Boc-cystine 15 was carried out using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) and 1-hydroxybenzotriazole (HOBt) in DMF. The column chromatography at this step was also avoided as the addition of crushed ice in the reaction mixture afforded crude product 16 (85% yield) which was found to be good enough to be used as it is. The reaction was carried out at up to 10 g scale and only a small amount was column purified to record the analytical data.
Various reagents are reported in literature for the cleavage of disulfide bond.35 Use of Bu3P reagent (50% solution in ethylacetate) was found to be more cost effective while working on large scale. The crucial point for the reaction was the addition of water (1 mL for 1 g batch). The reaction didn't proceed even after prolong reaction hours if enough quantity of water was not added into the reaction mixture. The solvent was evaporated under reduced pressure and the crude product 17 was used as it is for next step. The crude product 17 on reaction with synthon 5 in presence of K2CO3 in DMF solvent afforded compound 18 in excellent yield (90%). It is noteworthy to mention that no column chromatography was required for the purification of product 18 and the addition of crushed ice resulted in the formation of solid product in pure form.
Compound 18 was further treated with 70% acetic acid and stirred at room temperature for 16 h to furnish compound 19 in quantitative yield (96%).36 Compound 19 on O-palmitoylation using palmitoyl chloride and Et3N in the presence of catalytic amount of 4-dimethylaminopyridine (DMAP) afforded compound 20 which was purified by column chromatography.21 The global deprotection of Boc and tert-butyl ether functionalities was done by stirring the solution of compound 20 in TFA for 30 minutes at 25 °C. Evaporation of TFA in order to get pure product 21 was crucial and it was achieved by co-evaporation of the crude product with chloroform and dichloromethane till a white solid product was obtained which was confirmed by mass spectrometry. The solid TFA salt of amine was further treated with excess of di-tert-butyl dicarbonate and Et3N to furnish compound 22 ready for final coupling with tetralysine residue.37
The terapeptide H-[Lys(Boc)]4-OtBu 31 was synthesized in eight steps (Scheme 3) starting from commercially available carboxylic acid precursor Fmoc-Lys(Boc)-OH 23. The amine precursor of the appropriately protected lysine was synthesized in two steps as described by Pratesi et al.38 The general method for protection of carboxylic acid as tert-butyl esters involved the use of coupling reagents (EDCI and HOBt). In the present approach, a mixed anhydride was prepared using di-tert-butyl dicarbonate which on reaction with tert-butanol resulted in the formation of product 24.38 This protocol was found to be clean and high yielding. Further, compound 25 was obtained by treating the DMF solution of Fmoc-[Lys(Boc)]-OtBu 24 with 20% piperidine. The synthesis of H-[Lys(Boc)]2-OtBu 27 was obtained by the coupling reaction of reagent 23 and H-[Lys(Boc)]-OtBu 25 followed by the deprotection of Fmoc group as described. The formation of product 27 was confirmed by ESI-MS and 1H NMR.39
The formation of tripeptide H-[Lys(Boc)]3-OtBu 29 was achieved as mentioned above by using coupling reagents in DMF and following deprotection of Fmoc group. These reactions were reproducible and scalable in grams (2 g) with 74% and 86% yields, respectively. Similarly, the tetrapeptide H-[Lys(Boc)]4-OtBu 31 was synthesized through the coupling of starting precursor 23 with intermediate 29 and subsequent treatment with 20% piperidine with acceptable yields. The formation of tetrapeptide H-[Lys(Boc)]4-OtBu 31 was confirmed through mass spectrometric analysis. Ionic liquid mediated Fmoc removal40 as well as use of polymer-bound piperazine19 were the probable alternatives to avoid the column chromatography to remove the side products during this deprotection step but the column chromatography was found to be cost effective as well as simple during this process optimization.
The penultimate coupling of compound 22 with H-[Lys(Boc)]4-OtBu 31 (Scheme 4) using EDCI and HOBt coupling reagents in DMF resulted in the formation of an adduct 32 which on treatment with HCl in dioxane yielded the desired PAM2CSK4 2 in quantitative yield.
Conclusions
Overall, with a systematic reaction optimization and appropriate synthetic planning we have demonstrated a twenty step first complete synthesis of PAM2CSK4 which is scalable and will give an easy access to pharmacologically important TLR2 agonist capable of eliciting immune responses in both human and mice models. The complete procedure involved synthesis of two moieties, PAM2CS 3 (10 steps) and K4 tetrapeptide 4 (8 steps) and the final coupling and global deprotection to furnish PAM2CSK4 with 0.55% overall yield. For the cost effectiveness, the appropriately protected amino acids were synthesized by one step procedure from cheaply available cystine and serine. The present synthetic route will provide an easy access of PAM2CSK4 to explore its usefulness in the preparation of novel adjuvantic formulations.Experimental section
General
All the reactions were performed with commercially available reagents. The organic solvents were dried by standard procedures wherever necessary. The bulk solvents (hexane, DCM) were distilled before use. All reactions were monitored by thin-layer chromatography (TLC) carried out on Merck silica gel F254 aluminium sheets and visualized with different stains such as phosphomolybdic acid and ninhydrin solutions. All starting amino acid precursors were L enantiomers and purchased from Sigma aldrich. 1H NMR was recorded on Bruker Advance II 400 or 500 MHz or Jeol FT NMR 300 MHz. Mass analysis was performed with Waters, Q-TOF-Micromass (ESI-MS) Spectrometer.(R)-4-(Iodomethyl)-2,2-dimethyl-1,3-dioxolane (5)
To a solution of S-2,2-dimethyl-1,3-dioxolane-4-methanol 12 (1.0 g, 7.6 mmol) in DCM (10 mL) was added triphenylphosphine (2.4 g, 9.1 mmol), imidazole (1.5 g, 22.7 mmol), and iodine (2.5 g, 9.8 mmol). The reaction mixture was stirred at room temperature for 12 h. After the completion of reaction, DCM (100 mL) was added and the organic layer was washed with saturated sodium thiosulphate Na2S2O3 (100 mL × 2), brine (100 mL × 2), and dried over Na2SO4 and filtered. The organic solvent was then removed under vacuum and the residue was purified by flash column chromatography (2% EtOAc/hexane) to obtain compound (1.1 g, 63%). 1H NMR (400 MHz, CDCl3) δ 1.34 (s, 3H), 1.45 (s, 3H), 3.14 (m, 1H), 3.25 (dd, J = 9.8 Hz, 4.7 Hz, 1H), 3.78 (dd, J = 8.5 Hz, 5.4 Hz, 1H), 4.14 (dd, J = 8.6 Hz, 5.9 Hz, 1H), 4.27 (m, 1H). The 1H NMR was found to be consistent with that reported in the literature.28 For large scale (10 g), column purification was avoided by addition of warm 1.8 M methanolic solution of ZnCl2 (50 mL) into a DCM solution (10 mL) of the crude product which resulted in the formation of white shinning crystals of ZnCl2(Ph3PO)x31 after keeping the reaction mixture at room temperature for overnight. The reaction mixture was filtered and concentrated to yield pure product which was used as it for further transformations.tert-Butyl-O-(tert-butyl)-L-serinate (7)
To a suspension of L-serine (5 g, 0.047 mol) in tert-butylacetate (121 mL, 0.36 mol), 70% HCIO4 (4.6 mL, 76 mol) was added slowly by dropping funnel at 0 °C. After stirring at room temperature for 6 h, 2 N dil HCl (50 mL) was added to reaction mixture and further, basified to pH 9 by the addition of K2CO3 solution. The product was extracted using DCM (300 mL) and the organic layer was washed with, water (200 mL × 2), brine (200 mL × 2) and dried over Na2SO4, filtered and concentrated to afford (S)-tert-butyl 2-amino-3-tert-butoxypropanoate as colorless oil (8.26 g, 80%). 1H NMR (CDCl3, 300 MHz) δ 3.58 (m, 2H), 3.44 (m, 1H), 1.74 (s, 3H), 1.47 (s, 9H), 1.16 (s, 9H). 13C NMR (CDCl3, 100 MHz) δ 173.4, 80.9, 72.8, 63.9, 55.6, 28.0, 27.4. The 1H NMR and 13C NMR spectra were consistent with that reported in the literature.32(2R,2′R)-3,3′-Disulfanediylbis(2-((tert-butoxycarbonyl)amino)propanoicacid) (15)
Compound 15 was synthesized by a modified procedure as reported in the literature to avoid column chromatography on large scale. To a solution of L-cystine 14 (2 g, 8.32 mmol) in water (80 mL), triethylamine (3.5 mL, 24.97 mmol) was added followed by di-tert-butyl-dicarbonate (5.44 g, 24.97 mmol) and it was stirred at room temperature for 24 h. The reaction was quenched by adding 10% HCl solution (15 mL) at 0 °C. The product was precipitated out as a white solid which was filtered and washed with cold distilled water to obtain pure compound 15 (2.7 g, 74%). 1H NMR (CDCl3, 300 MHz) δ 5.6 (m, 1H), 4.5 (m, J = 4.8 Hz, 2H), 3.24 (m, 4H), 1.45 (s, 18H). The 1H NMR was consistent with that reported in the literature.33Di-tert-butyl-2,2′-(((2R,2′R)-3,3′-disulfanediylbis(2-((tert-butoxycarbonyl)amino)propanoyl))bis(azanediyl))(2S,2′S)-bis(3-(tert-butoxy)propanoate) (16)
To a solution of Nα,Nα′-di-Boc-cystine 15 (0.69 g, 1.58 mmol) in anhydrous DMF were added H-Ser(tBu)-OtBu·HCl 7 (0.84 g, 3.31 mmol), HOBt (0.513 g, 3.31 mmol), and Et3N (0.46 mL, 3.31 mmol). The reaction mixture was stirred at 0 °C for 30 min, followed by addition of EDCI (0.24 g, 1.58 mmol) at 0 °C. The reaction mixture was allowed to come to room temperature and stirred for 6 h. After completion of reaction, the solvent was evaporated under reduced pressure and residue obtained was dissolved in ethyl acetate (250 mL). It was washed with water (150 mL × 2), brine (150 mL × 1). The organic layer was dried over anhydrous Na2SO4, filtered, and evaporated under reduced pressure to obtain crude product which was purified using column chromatography (50% EtOAc/hexane) to obtain compound 16 (1.12 g, 85%). 1H NMR (500 MHz, CDCl3) δ 7.35 (s, 2H), 5.69 (s, 2H), 4.71–4.50 (m, 4H), 3.77 (dd, J = 8.8, 3.4 Hz, 2H), 3.55 (dd, J = 8.8, 3.4 Hz, 2H), 3.18–3.06 (m, 4H), 1.46 (s, 36H), 1.14 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 170.03, 168.95, 155.39, 81.77, 80.16, 73.35, 62.08, 53.54, 43.22, 28.42, 28.02, 27.35. MS (ESI) calculated for [M + Na]+ C38H70N4NaO12S2+ m/z 861.43, found 861.44. For large scale, compound 16 was used without column chromatography after precipitation as discussed in the result and discussion section.tert-Butyl-N-(N-(tert-butoxycarbonyl)-S-(((R)-2,2-dimethyl-1,3-dioxolan-4-yl)methyl)-L-cysteinyl)-O-(tert-butyl)-L-serinate (18)
To a solution of compound 16 (1 g, 1.29 mmol) in DCM (20 mL) and water (1 mL), tributylphosphine (50% solution in EtOAc, 964 μL, 3.86 mmol) was added and the reaction mixture was stirred at room temperature for 30 minutes. After the completion of the reaction, the solvent was removed under reduced pressure to obtain the crude product 17 (720 mg, 74%). The small amount of crude product 17 was column purified for the analysis. 1H NMR (500 MHz, CDCl3) δ 6.90 (d, J = 7.9 Hz, 1H), 5.50 (d, J = 6.8 Hz, 1H), 4.54 (dt, J = 8.0, 2.8 Hz, 1H), 4.37 (s, 1H), 3.79 (dd, J = 8.8, 2.9 Hz, 1H), 3.55 (dd, J = 8.8, 3.0 Hz, 1H), 3.08 (ddd, J = 13.9, 8.0, 4.2 Hz, 1H), 2.75 (ddd, J = 13.9, 10.2, 6.3 Hz, 1H), 1.77 (t, J = 8.8 Hz, 1H), 1.46 (s, 18H), 1.14 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 169.55, 168.95, 155.20, 82.11, 80.32, 73.22, 61.81, 55.57, 53.52, 28.30, 28.01, 27.47, 27.35. To the solution of crude compound 17 (700 mg, 1.67 mmol) in DMF (10 mL) were added K2CO3 (346 mg, 2.51 mmol) and compound 5 (1.01 g, 4.17 mmol). The reaction mixture was stirred at room temperature for 2 h. After completion of the reaction, reaction mixture was poured onto crushed ice with stirring and product was extracted with DCM (250 mL), followed by washings with water (150 mL × 2), brine (150 mL × 2). The organic layer was finally concentrated to obtain crude product which was purified using column chromatography (25% EtOAc/hexane) to obtain compound 18 (800 mg, 90%). 1H NMR (500 MHz, CDCl3) δ 7.06 (d, J = 8.0 Hz, 1H), 5.50 (s, 1H), 4.54 (dt, J = 8.0, 2.9 Hz, 1H), 4.35–4.26 (m, 2H), 4.10 (dd, J = 8.3, 6.1 Hz, 1H), 3.79 (dd, J = 8.8, 2.9 Hz, 1H), 3.70 (dd, J = 8.3, 6.6 Hz, 1H), 3.53 (dd, J = 8.8, 3.0 Hz, 1H), 3.05 (dd, J = 13.9, 5.9 Hz, 1H), 2.94 (dd, J = 13.9, 6.3 Hz, 1H), 2.82 (dd, J = 13.5, 6.2 Hz, 1H), 2.71 (dd, J = 13.5, 6.1 Hz, 1H), 1.46 (d, J = 1.7 Hz, 18H), 1.44 (s, 3H), 1.36 (s, 3H), 1.15 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 170.18, 168.91, 155.28, 109.67, 81.94, 80.17, 75.59, 73.13, 68.74, 61.99, 54.07, 53.49, 35.56, 35.32, 28.30, 28.01, 27.35, 26.87, 25.56. MS (ESI) calculated for [M + Na]+ C25H46N2NaO8S+ m/z 557.28, found 558.26.tert-Butyl-N-(N-(tert-butoxycarbonyl)-S-((R)-2,3-dihydroxypropyl)-L-cysteinyl)-O-(tert-butyl)-L-serinate (19)
The compound 18 (2.9 g) was dissolved in 70% acetic acid (30 mL, AcOH:water = 7:3) and the reaction mixture was stirred at room temperature for 16 h. After completion of the reaction, the reaction mixture was diluted with EtOAc (350 mL) and washed with 10% sodium bicarbonate (200 mL × 2), water (200 mL × 2) and brine (200 mL) and organic layer was concentrated under reduced pressure to yield crude product which was purified using column chromatography (5% MeOH/DCM) to obtain compound 19 (2.5 g, 96%). 1H NMR (500 MHz, CDCl3) δ 7.23 (d, J = 7.7 Hz, 1H), 5.63 (d, J = 8.1 Hz, 1H), 4.57 (dt, J = 8.1, 2.9 Hz, 1H), 4.42 (d, J = 5.8 Hz, 1H), 3.89–3.83 (m, 1H), 3.81–3.78 (m, 1H), 3.70 (dd, J = 11.3, 3.6 Hz, 2H), 3.60 (dd, J = 11.3, 6.2 Hz, 1H), 3.55 (dd, J = 8.9, 3.0 Hz, 1H), 3.00 (dd, J = 14.1, 5.6 Hz, 1H), 2.91 (dd, J = 14.1, 6.7 Hz, 1H), 2.78 (m, 2H), 1.47 (s, 9H), 1.45 (s, 9H), 1.16 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 170.23, 169.20, 155.56, 82.26, 80.36, 73.34, 71.10, 65.23, 62.02, 53.49, 35.93, 28.30, 28.02, 27.34. MS (ESI) calculated for [M + Na]+ C22H37N2NaO8S+ m/z 517.25, found 517.23. For large scale, compound 19 was used further without any column purification.
((R)-3-((R)-2-(tert-Butoxycarbonylamino)-3-((S)-1,3-di-tert-butoxy-1-oxopropan-2-ylamino)-3-oxopropylthio)propane-1,2-diyldipalmitate) (20)
To a solution of compound 19 (650 mg, 1.31 mmol) in anhydrous DCM (15 mL) were added Et3N (733 μL, 5.26 mmol) and a catalytic amount of DMAP (20 mol%). The reaction mixture was stirred at 0 °C for 30 minutes. Palmitoyl chloride (1.2 mL, 3.94 mmol) was then added at 0 °C, followed by stirring at room temperature for 12 h. After completion of the reaction, the reaction mixture was thoroughly washed with water (250 mL × 2), saturated sodium bicarbonate solution (250 mL × 2), brine (200 mL × 1). The organic layer was dried over anhydrous Na2SO4, filtered and, evaporated under reduced pressure to obtain crude product which was purified using column chromatography (20% EtOAc/hexane) to obtain compound 20 (880 mg, 68%). 1H NMR (500 MHz, CDCl3) δ 7.04 (d, J = 8.0 Hz, 1H), 5.40 (d, J = 6.1 Hz, 1H), 5.18 (dd, J = 5.7, 3.7 Hz, 1H), 4.53 (dt, J = 8.1, 2.9 Hz, 1H), 4.33 (dd, J = 11.9, 3.5 Hz, 2H), 4.17 (dd, J = 11.9, 5.9 Hz, 1H), 3.79 (dd, J = 8.8, 2.9 Hz, 1H), 3.53 (dd, J = 8.8, 3.0 Hz, 1H), 2.96 (d, J = 6.3 Hz, 2H), 2.82 (d, J = 6.0 Hz, 2H), 2.32 (ddd, J = 12.3, 6.7, 3.0 Hz, 4H), 1.67–1.56 (m, 5H), 1.46 (d, J = 3.5 Hz, 18H), 1.27 (d, J = 15.1 Hz, 48H), 1.15 (s, 9H), 0.88 (t, J = 7.0 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 173.50, 173.23, 170.19, 169.03, 155.36, 82.06, 80.28, 73.26, 70.45, 63.63, 62.09, 54.12, 53.64, 35.77, 34.43, 34.23, 33.28, 32.07, 29.85, 29.81, 29.66, 29.51, 29.46, 29.45, 29.29, 29.28, 28.43, 27.48, 25.04, 25.02, 22.84, 14.27. MS (ESI) calculated for [M + Na]+ C54H102N2NaO10S+, m/z 993.71, found 993.19.S-((R)-2,3-Bis(palmitoyloxy)propyl)-N-(tert-butoxycarbonyl)-L-cysteinyl-L-serine (22)
The compound 20 (500 mg, 0.53 mmol) was stirred with trifluoroacetic acid (TFA, 8 mL) for 30 minutes at 25 °C, followed by purging nitrogen gas. After that remaining TFA was co-evaporated with CHCl3 (5 mL × 3) and DCM (5 mL × 2) in order to get solid crude compound which was purified by column chromatography (10% MeOH/DCM) to get compound 21 (350 mg, 89%). MS (ESI) calculated for [M + H]+ C41H78N2O8S m/z 758.54, found 758.28. To a solution of compound 21 (286 mg, 0.32 mmol) in DCM, di-tert-butyl dicarbonate (246 mg, 1.31 mmol) Et3N (133 μL, 0.92 mmol) were added and the reaction mixture was stirred for 4 h. The reaction mixture was diluted with DCM (300 mL) and organic layer was sequentially washed with water (250 mL × 2), saturated sodium bicarbonate solution (250 mL × 2), brine (200 mL × 1). The organic layer was dried over anhydrous sodium sulfate, filtered and evaporated under reduced pressure to obtain the crude product which was purified using column chromatography (5% MeOH/DCM) to afford compound 22 (188 mg, 67%). 1H NMR (500 MHz, CDCl3) δ 12.17–11.77 (m, 1H), 7.59–7.48 (m, 1H), 5.67–5.54 (m, 1H), 5.20–5.13 (m, 1H), 4.63–4.53 (m, 1H), 4.44–4.36 (m, 1H), 4.32 (s, 1H), 4.14 (s, 1H), 4.09–4.01 (m, 1H), 3.96–3.82 (m, 1H), 3.10 (s, 1H), 3.06–2.88 (m, 2H), 2.76 (s, 2H), 2.29 (d, J = 7.4 Hz, 4H), 1.65–1.55 (m, 4H), 1.43 (s, 9H), 1.21 (s, 48H), 0.86 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 173.89, 173.74, 171.04, 156.02, 80.96, 77.41, 77.16, 76.91, 70.53, 63.84, 62.82, 55.31, 54.04, 45.82, 35.16, 34.50, 34.25, 32.91, 32.08, 29.87, 29.83, 29.70, 29.68, 29.53, 29.48, 29.32, 29.29, 28.42, 25.06, 25.02, 22.85, 14.28, 8.69. MS (ESI) calculated for [M + Na]+ C46H86N2NaO10S+ m/z 881.58, found 881.58.tert-Butyl-N2-(((9H-fluoren-9-yl)methoxy)carbonyl)-N6-(tert-butoxycarbonyl)-L-lysinate (Fmoc-Lys(Boc)-OtBu) (24)
The compound 24 was synthesized using precursor Fmoc-Lys(Boc)-OH 23 (5.0 g, 10.7 mmol) using a reported protocol.38 Yield (3.91 g, 70%). 1H NMR (400 MHz, CDCl3); δ 7.76 (d, J = 7.5 Hz, 2H), 7.60 (d, J = 7.3 Hz, 2H), 7.40 (t, J = 7.5 Hz, 2H), 7.31 (t, J = 7.5 Hz, 2H), 5.36 (d, J = 8 Hz, 1H), 4.37–4.42 (m, 2H), 4.20–4.27 (m, 2H), 3.10 (t, J = 6.6 Hz, 2H), 1.77–1.86 (m, 4H), 1.47 (s, 9H), 1.43 (s, 11H).tert-Butyl-N6-(tert-butoxycarbonyl)-L-lysinate (H-Lys(Boc)-OtBu) (25)
The compound 25 (4.0 g, 7.62 mmol) was synthesized as reported in the literature38 and confirmed by mass spectrometric analysis. Yield (1.75 g, 76%). MS (ESI) calculated for [M + H]+ C15H31N2O4+ m/z 303.22, found 303.53.tert-Butyl-N2-(N2-(((9H-fluoren-9-yl)methoxy)carbonyl)-N6-(tert-butoxycarbonyl)-L-lysyl)-N6-(tert-butoxycarbonyl)-L-lysinate (Fmoc-Lys(Boc)-Lys(Boc)-OtBu) (26)
The compound 26 (1.6 g, 5.2 mmol) was obtained using similar procedure as reported and by using pyridine instead of N,N-diisopropylethylamine (DIPEA).39 Yield (3.08 g, 87%). 1H NMR (400 MHz, CDCl3) δ 7.75 (d, J = 7.5 Hz, 2H), 7.59 (d, J = 7.4 Hz, 2H), 7.39 (t, J = 7.4 Hz, 2H), 7.30 (m, 2H), 6.57 (bs, 1H), 5.59 (bs, 1H), 4.67 (bs, 2H), 4.37–4.46 (m, 3H), 4.11–4.23 (m, 2H), 3.05–3.11 (m, 4H), 1.83 (m, 2H), 1.71 (m, 4H), 1.43 (m, 32H), 1.25 (m, 2H), 1.19–1.21 (m, 2H).tert-Butyl-N6-(tert-butoxycarbonyl)-N2-(N6-(tert-butoxycarbonyl)-L-lysyl)-L-lysinate (H-Lys(Boc)-Lys(Boc)-OtBu) (27)
The deprotection of Fmoc-Lys(Boc)-Lys(Boc)-OtBu 26 (500 mg, 0.66 mmol) was carried out as described for compound 24 to afford compound 27.39 Yield (334.8 mg, 95%).1H NMR (400 MHz, CDCl3) δ 7.71 (d, J = 7.0 Hz, 1H), 4.69 (bs, 2H), 4.42–4.47 (m, 1H), 3.37–3.40 (m, 1H), 3.09–3.12 (m, 4H), 1.80 (m, 4H), 1.45–1.75 (m, 8H), 1.45 (s, 9H), 1.42 (s, 18H). MS (ESI) calculated for [M + H]+ C26H51N4O7 m/z 531.37, found 531.40.tert-Butyl-N2-(N2-(N2-(((9H-fluoren-9-yl)methoxy)carbonyl)-N6-(tert-butoxycarbonyl)-L-lysyl)-N6-(tert-butoxycarbonyl)-L-lysyl)-N6-(tert-butoxycarbonyl)-L-lysinate (Fmoc-Lys(Boc)-Lys(Boc)-Lys(Boc)-OtBu) (28)
H-Lys(Boc)-Lys(Boc)-OtBu 27 (400 mg, 0.75 mmol) and Fmoc-Lys-OH 23 (317.8 mg, 0.678 mmol) were dissolved in 5 mL of DMF at 0 °C at inert atmosphere. HOBt (51 mg, 0.376 mmol) and pyridine (0.12 mL, 1.5 mmol) were further added to the reaction mixture at 0 °C. After 10 minutes stirring, EDCI (216.5 mg, 1.13 mmol) was added to the reaction mixture and reaction was allowed to warm to room temperature and stirred for 6 h. The completion of the reaction was monitored by TLC. The reaction mixture was dissolved in water (100 mL) and EtOAc (200 mL) and the organic phase was washed successively with 5% citric acid (50 mL × 2), NaHCO3 (50 mL × 2) and brine (50 mL × 2). The crude product obtained was purified by column chromatography (2% MeOH/DCM) to obtain compound 28 (547 mg, 74%). 1H NMR (400 MHz, CDCl3) δ 7.74–7.76 (d, J = 7.5 Hz, 2H), 7.59–7.60 (d, J = 7.0 Hz, 2H), 7.38 (t, J = 7.4 Hz, 2H), 7.30 (t, J = 7.2 Hz, 2H), 6.94 (m, 1H), 6.83 (m, 1H), 5.81 (m, 1H), 4.93 (m, 1H), 4.83 (m, 1H), 4.39 (m, 3H), 4.20 (t, J = 7.0 Hz, 2H), 3.05 (m, 5H), 2.06 (m, 2H), 1.84 (m, 3H), 1.65 (m, 3H), 1.42 (m, 44H). 13C NMR δ 171.23, 156.31, 156.19, 141.26, 127.72, 127.11, 125.14, 119.96, 82.10, 79.10, 77.25, 67.17, 54.85, 52.68, 47.10, 40.11, 39.94, 39.78, 32.06, 31.76, 29.48, 28.47, 28.01, 22.53, 22.32. MS (ESI) calculated for [M + Na]+ C52H80N6NaO12+ m/z 1003.57, found 1003.54.tert-Butyl-N6-(tert-butoxycarbonyl)-N2-(N6-(tert-butoxycarbonyl)-N2-(N6-(tert-butoxycarbonyl)-L-lysyl)-L-lysyl)-L-lysinate (H-Lys(Boc)-Lys(Boc)-Lys(Boc)-OtBu) (29)
Fmoc-Lys(Boc)-Lys(Boc)-Lys(Boc)-OtBu 28 (450 mg, 0.458 mmol) was dissolved in a solution of 20% of piperidine in DMF (3 mL) and stirred for 1 h. After completion of reaction, the solvent was evaporated and the crude product obtained was purified by column chromatography (10% MeOH/DCM) to obtain compound 29 (300 mg, 86%). MS (ESI) calculated for [M + H]+ C37H71N6O10 m/z 759.52, found 759.24.tert-Butyl-(2R,5S)-5-((2R,5S)-5-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-9-((tert-butoxycarbonyl)amino)-2-(4-((tert-butoxycarbonyl)amino)butyl)-4-oxononanamido)-9-((tert-butoxycarbonyl)amino)-2-(4-((tert-butoxycarbonyl)amino)butyl)-4-oxononanoate (Fmoc-Lys(Boc)-Lys(Boc)-Lys(Boc)-Lys(Boc)-OtBu) (30)
NH2-Lys(Boc)-Lys(Boc)-Lys(Boc)-OtBu 29 (300 mg, 0.40 mmol) and Fmoc-Lys-OH 23 (166.7 mg, 0.36 mmol) were dissolved in 10 mL of DMF at 0 °C under nitrogen atmosphere. HOBt (26.7 mg, 0.197 mmol) and pyridine (63.7 μL, 0.79 mmol) were further added to the reaction mixture under cooling condition. After 10 minutes stirring, EDCI (113.6 mg, 0.59 mmol) was added to the reaction mixture and reaction was allowed to warm to room temperature and stirred for 6 h. The completion of the reaction was monitored by TLC. The reaction mixture was dissolved in water (150 mL) and EtOAc (200 mL) and the organic phase was washed successively with 5% citric acid (50 mL × 2), NaHCO3 (50 mL × 2) and brine (50 mL × 2). The crude product obtained was purified by column chromatography on 60–120 mesh silica gel in 4% MeOH/DCM to obtain compound 30 (301 mg, 63%). 1H NMR (400 MHz, CDCl3) δ 9.08 (bs, 1H), 8.54 (bs, 1H), 7.75 (d, J = 7.5 Hz, 2H), 7.59–7.61 (d, J = 6.4 Hz, 2H), 7.38 (t, J = 7.4 Hz, 2H), 7.29 (m, 2H), 6.98 (m, 1H), 6.76 (m, 1H), 5.93–6.08 (m, 2H), 5.37 (m, 1H), 4.96 (m, 1H), 4.82–4.86 (m, 1H), 4.32–4.56 (m, 6H), 4.19–4.22 (m, 2H), 2.88–3.07 (m, 9H), 1.85 (m, 6H), 1.59–1.71 (m, 7H), 1.43 (m, 67H). 13C NMR (100 MHz, CDCl3) δ 171.34, 156.44, 141.39, 127.78, 127.19, 125.37, 120.0, 60.55, 47.32, 28.62, 28.13, 22.83, 21.21, 14.34. MS (ESI) calculated for [M + Na]+ C63H100N8NaO15+ m/z 1231.72, found 1231.63.tert-Butyl-(2R,5S)-5-((2R,5S)-5-amino-9-((tert-butoxycarbonyl)amino)-2-(4-((tert-butoxycarbonyl)amino)butyl)-4-oxononanamido)-9-((tert-butoxycarbonyl)amino)-2-(4-((tert-butoxycarbonyl)amino)butyl)-4-oxononanoate (H-Lys(Boc)-Lys(Boc)-Lys(Boc)-Lys(Boc)-OtBu) (31)
Fmoc-Lys(Boc)-Lys(Boc)-Lys(Boc)-Lys(Boc)-OtBu 30 (300 mg, 0.248 mmol) was dissolved in a solution of 20% of piperidine in DMF (2 mL) and stirred for 1 h. The solvent was evaporated and the crude product obtained was purified by column chromatography on 60–120 mesh silica gel in 15% MeOH/DCM to obtain compound 31 (150 mg, 61%). MS (ESI) calculated for [M + H]+ C48H91N8O13+ m/z 987.67, found 987.65.S-(2,3-Bis(palmitoyloxy)propyl)-cysteinyl-seryl-lysyl-lysyl-lysyl-lysine (2)
To a solution of compound 22 (100 mg, 0.12 mmol) in anhydrous DMF were added H-[Lys(Boc)]4-OtBu 31 (126 mg, 0.13 mmol), HOBt (16 mg, 0.12 mmol), and Et3N (34 μL, 0.24 mmol). The reaction mixture was stirred at 0 °C for 30 min, followed by addition of EDCI (69.0 mg, 0.36 mmol) at 0 °C. The reaction mixture was further stirred at room temperature for 6 h, followed by evaporation of the solvent under reduced pressure. The residue was then dissolved in ethyl acetate and washed with 5% critic acid (100 mL × 2), NaHCO3 (100 mL × 2) and brine (100 mL × 2). The organic solvent was dried over anhydrous sodium sulfate, filtered, and evaporated under reduced pressure to obtain the crude product which was purified using column chromatography (50% EtOAc/hexane) to obtain compound 32 (145 mg, 68%). The compound 32 (50 mg) was treated with HCl in dioxane solution and stirred for 2 h to obtain compound 2 (28.5 mg, 82%). 1H NMR (500 MHz, MeOD) δ 5.30 (ddd, J = 8.0, 6.5, 2.8 Hz, 2H), 4.57 (t, J = 5.7 Hz, 2H), 4.47 (dd, J = 12.0, 2.7 Hz, 2H), 4.37 (dddd, J = 19.6, 14.4, 9.3, 5.0 Hz, 8H), 4.24 (dd, J = 9.0, 5.1 Hz, 2H), 4.17 (dd, J = 12.0, 6.7 Hz, 2H), 3.96 (dd, J = 11.0, 5.4 Hz, 2H), 3.84 (dd, J = 11.0, 6.0 Hz, 2H), 3.75–3.73 (m, 1H), 3.69–3.65 (m, 2H), 3.60–3.57 (m, 1H), 3.28 (d, J = 5.1 Hz, 2H), 2.99 (dt, J = 13.8, 5.8 Hz, 18H), 2.89 (dd, J = 14.6, 9.0 Hz, 2H), 2.82 (dd, J = 14.3, 8.3 Hz, 2H), 2.35 (dt, J = 13.3, 7.4 Hz, 8H), 1.98–1.84 (m, 9H), 1.83–1.66 (m, 24H), 1.66–1.44 (m, 25H), 1.39–1.15 (m, 91H), 0.90 (t, J = 7.0 Hz, 12H). 13C NMR (126 MHz, MeOD) δ 175.02, 174.98, 174.91, 174.21, 174.12, 174.06, 172.33, 168.94, 73.57, 72.45, 71.33, 65.23, 63.19, 62.17, 56.77, 55.07, 54.68, 54.52, 53.41, 53.30, 40.60, 40.58, 40.52, 40.47, 35.23, 34.99, 34.00, 33.11, 33.00, 32.29, 32.17, 31.78, 30.85, 30.81, 30.75, 30.72, 30.55, 30.52, 30.27, 30.26, 27.99, 27.90, 27.85, 26.14, 26.07, 23.89, 23.86, 23.76, 23.64, 14.47. MS (ESI) calculated for [M + H]+ C65H127N10O12S+ m/z 1271.93, found 1271.92.Conflicts of interest
There are no conflicts to declare.Acknowledgements
DBS is thankful to UGC New Delhi for start-up research grant and DBT New Delhi for the award of Ramalingaswami Fellowship. The support from UGC-CAS, DST-PURSE-II, DST-SAIF and Panjab University Development fund is gratefully acknowledged. AK is thankful to UGC, New Delhi for the award of Junior Research Fellowship. MTP is thankful to DST for the award of Women Scientist Scheme-A (WOS-A).References
- T. H. Mogensen, Clin. Microbiol. Rev., 2009, 22, 240 CrossRef CAS PubMed.
- T. Kawasaki and T. Kawai, Front. Immunol., 2014, 5, 1 CAS.
- D. Werling and T. W. Jungi, Vet. Immunol. Immunopathol., 2003, 91, 1 CrossRef CAS PubMed.
- A. G. Dalgleish, Hum. Vaccines Immunother., 2014, 10, 3369 CrossRef PubMed.
- T. Kawai and S. Akira, Nat. Immunol., 2010, 11, 373 CrossRef CAS PubMed.
- A. O. Aliprantis, R.-B. Yang, M. R. Mark, S. Suggett, B. Devaux, J. D. Redolf, G. R. Klimpel, P. Godowski and A. Zychlinsky, Science, 1999, 285, 736 CrossRef CAS PubMed.
- (a) M. S. Jin, S. E. Kim, J. Y. Heo, M. E. Lee, H. M. Kim, S. G. Paik, H. Lee and J. O. Lee, Cell, 2007, 130, 1071 CrossRef CAS PubMed; (b) J. Y. Kang, X. Nan, M. S. Jin, S. J. Youn, Y. H. Ryu, S. Mah, S. H. Han, H. Lee, S. G. Paik and J. O. Lee, Immunity, 2009, 31, 873 CrossRef CAS PubMed.
- M. Murata, Cancer Sci., 2008, 99, 1435 CrossRef CAS PubMed.
- N. Isambert, P. Fumoleau, C. Paul, C. Ferrand, S. Zanetta, J. Bauer, K. Ragot, G. Lizard, J. Jeanin and M. Bardou, BMC Cancer, 2013, 13, 172 CrossRef CAS PubMed.
- M. A. Moreno-Eutimio, A. Tenorio-Calvo, R. Pastelin-Palacios, C. Perez-Shibayama, C. Gil-Cruz, R. Lopez-Santiago, I. Baeza, M. Fernandez-Mora, L. Bonifaz, A. Isibasi, E. Calva and C. Lopez-Macias, Immunology, 2013, 139, 459 CrossRef CAS PubMed.
- A. Halliday, J. D. Turner, A. Guimarães, P. A. Bates and M. J. Taylor, Parasites Vectors, 2016, 9, 96 CrossRef PubMed.
- T. J. Powell, WO Pat., 2013148426A1, 2013.
- A. Maruyama, H. Shime, Y. Takeda, M. Azuma, M. Matsumoto and T. Seya, Biochem. Biophys. Res. Commun., 2015, 457, 445 CrossRef CAS PubMed.
- M. C. Nawijn, A. C. Motta, R. Gras, S. Shirinbak, H. Maazi and A. J. M. Oosterhout, PLoS One, 2013, 8, e55307 CAS.
- J. R. Slingluff, L. Craig and I. S. Mauldin, WO Pat., 201507003A1, 2015.
- U. Buwitt-Beckmann, H. Heine, K. H. Wiesmuller, G. Jung, R. Brock and A. J. Ulmer, FEBS J., 2005, 272, 6354 CrossRef CAS PubMed.
- W. Zeng, S. Ghosh, Y. F. Lau, L. E. Brown and D. C. Jackson, J. Immunol., 2002, 169, 4905 CrossRef.
- (a) J. W. Metzger, A. G. Beck-Sickinger, M. Loleit, M. Eckert, W. G. Bessler and G. J. Jung, Pept. Sci., 1995, 3, 184 CrossRef PubMed; (b) P. F. Muhlradt, M. Kiess, H. Meyer, R. Sussmuth and G. Jung, J. Exp. Med., 1997, 185, 1951 CrossRef CAS PubMed; (c) P. F. Muhlradt, M. Kiess, H. Meyer, R. Sussmuth and G. Jung, Infect. Immun., 1998, 66, 4804 CAS.
- D. B. Salunke, S. W. Connelly, N. M. Shukla, A. R. Hermanson, L. M. Fox and S. A. David, J. Med. Chem., 2013, 56, 5885 CrossRef CAS PubMed.
- D. B. Salunke, X. Guo and S. A. David, US Pat., 20150337009, 2015.
- W. Wu, R. Li, S. S. Malladi, H. J. Warshakoon, M. R. Kimbrell, M. W. Amolins, R. Ukani, A. Datta and S. A. David, J. Med. Chem., 2010, 53, 3198 CrossRef CAS PubMed.
- M. R. Kimbrell, H. Warshakoon, J. R. Cromer, S. Malladi, J. D. Hood, R. Balakrishna, T. A. Scholdberg and S. A. David, Immunol. Lett., 2008, 118, 132 CrossRef CAS PubMed.
- J. D. Hood, H. J. Warshakoon, M. R. Kimbrell, N. M. Shukla, S. S. Malladi, X. Wang and S. A. David, Hum. Vaccines, 2010, 6, 322 CrossRef CAS.
- M. Azuma, R. Sawahata, Y. Akao, E. Takashi, S. Yamazaki, M. Matsumoto, M. Hashimoto, K. Fukase, Y. Fujimoto and S. Teya, PLoS One, 2010, 5, e12550 Search PubMed.
- J. C. Farges, F. Carrouela, J.-F. Keller, C. Baudouin, P. Msika, F. Bleicher and M.-J. Staquet, Immunobiology, 2011, 216, 513 CrossRef CAS PubMed.
- A. C. D. Salyer, G. Caruso, K. K. Khetani, L. M. Fox, S. S. Malladi and S. A. David, PLoS One, 2016, 11, e0149848 Search PubMed.
- L. W. Hamley, S. Kirkham, A. Dehsorkhi, V. Castelletto, M. Reza and J. Ruokolainen, Chem. Commun., 2014, 50, 15948 RSC.
- http://www.invivogen.com/pam2csk4.
- N. Martin and E. J. Thomas, Org. Biomol. Chem., 2012, 10, 7952 CAS.
- P. J. Garegg, T. Regberg, J. Stawinski and R. Strornberg, J. Chem. Soc., Perkin Trans. 2, 1987, 11, 271 RSC.
- D. C. Batesky, M. J. Goldfogel and D. J. Weix, J. Org. Chem., 2017, 82, 9931 CrossRef CAS PubMed.
- L. Sambri, G. Bartoli, G. Bencivenni and R. Dalpozzo, Curr. Org. Synth., 2012, 9, 137 CrossRef CAS.
- P. Plastina, A. Fazio, M. Attya, G. Sindona and B. Gabriele, Nat. Prod. Res., 2012, 26, 1799 CrossRef CAS PubMed.
- D. B. Salunke, N. M. Sukhla, Y. Euna, B. M. Crall, R. Balakrishna, S. S. Malladi and S. A. David, J. Med. Chem., 2012, 55, 3353 CrossRef CAS PubMed.
- (a) Y. Hao, M. Zhang, J. He and P. Ni, Langmuir, 2012, 28, 6448 CrossRef CAS PubMed; (b) S. H. Lee and H. Kohn, Heterocycles, 2003, 60, 47 CrossRef CAS; (c) R. D. Besten, S. Hardeer and L. J. Brandsma, Organomet. Chem., 1990, 385, 153 CrossRef.
- G. Agnihotri, B. M. Crall, T. C. Lewis, T. P. Day, R. Balakrishna, H. J. Warshakoon, S. S. Malladi and S. A. David, J. Med. Chem., 2011, 54, 8148 CrossRef CAS PubMed.
- S. A. David and N. M. Shukla, US Pat., 201202944885, 2012.
- (a) A. Pratesi, M. Ginanneschi, F. Melani, M. Chinol, A. Carollo, G. Paganelli, M. Lumini, M. Bartoli, M. Frediani, L. Rosi, G. Petrucci, L. Messori and A. M. Papini, Org. Biomol. Chem., 2015, 13, 3988 RSC; (b) S. Banerjee, W. J. Wiggins, J. L. Geoghegan, C. T. Anthony, E. A. Woltering and D. S. Masterson, Org. Biomol. Chem., 2013, 11, 6307 RSC; (c) C. Kim, T. Young, Y. Cao, J. Ma, M. Kim, S. Pinkerton and P. G. Schultz, WO Pat., 2015057852, 2015.
- J. T. Herpt, M. C. A. Stuart, W. R. Browne and B. L. Feringa, Chem. - Eur. J., 2014, 20, 3077 CrossRef PubMed.
- M. L. Di Gioia, P. Costanzo, A. De Nino, L. Maiuolo, M. Nardi, F. Olivitod and A. Procopiod, RSC Adv., 2017, 7, 36482 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Activity profiling table for PAM2CSK4 with references; 1H NMR, 13C NMR and MS spectra of intermediates and final product PAM2CSK4. See DOI: 10.1039/c8ra01387j |
| This journal is © The Royal Society of Chemistry 2018 |