
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDifference in the deactivation of Au catalysts during ethanol transformation when supported on ZnO and on TiO2†
M. V. Moralesa,
E. Asedegbega-Nieto *a,
E. Castillejos-Lópeza,
B. Bachiller-Baezab and
A. Guerrero-Ruiza
*a,
E. Castillejos-Lópeza,
B. Bachiller-Baezab and
A. Guerrero-Ruiza
aDepartment of Inorganic Chemistry and Chemical Engineering, UNED, Madrid, Spain. E-mail: easedegbega@ccia.uned.es
bInstitute of Catalysis and Petrochemistry, CSIC, Madrid, Spain
First published on 15th February 2018
Abstract
Au nanoparticles of different sizes were supported by the deposition–precipitation method on two metal oxides: ZnO and TiO2. The resulting catalysts were tested in the ethanol catalytic transformation reaction. Both metal oxide support materials exerted a different influence on the achieved Au particle size as well as on the behavior of the subsequent catalyst, with regard to their initial conversion values, product distribution and stability. While TiO2 favors the formation of smaller nanoparticles, ZnO offers larger Au particle sizes when prepared under similar conditions. At the same time, TiO2 produced catalysts which displayed higher initial conversions in comparison with AuZnO catalysts, even when observing catalysts of each series with similar particle sizes. At the same time, catalysts supported on ZnO exhibited higher resistance to deactivation caused by coke formation. These results were evidenced employing different characterization techniques on both used and fresh catalyst samples. The decline in deactivation was generally accompanied by an increase in the carbon content on the catalyst's surface.
Introduction
Oxidation, dehydration and dehydrogenation reactions involving ethanol are widely studied due to the valuable chemicals obtained in these processes.1–3 An important example is the dehydrogenation of ethanol to produce acetaldehyde which is an intermediate for the synthesis of higher C4 hydrocarbons.4 On the other hand, simple probe molecule reaction tests, such as ethanol transformation, aid in investigation purposes, looking into the behavior of catalysts in terms of the optimum nanoparticle size of the active phase, metal-support interaction, causes of deactivation, etc.5,6 Among the catalysts studied, the use of transition metal oxides has greatly increased the number of publications in this field when employed as supports or as active catalysts on their own. In this latter case, properties such as acid–base, redox and others, make these materials interesting candidates. Acid–base properties have been studied via various techniques such as chemisorption of probe molecules, microcalorimetry and IR spectroscopy,7,8 among others. Classification of metal oxides as strictly acidic or basic is not always straightforward. For example, ZrO2 and TiO2 are said to be significantly basic with medium to high Lewis and weak Brønsted acid sites.9–11 However, as is reported in these first two mentioned publications, these properties can be adjusted by combining with other metal oxides like CeO2 and La2O3. As for γ-alumina, it possesses various types of Lewis and Brønsted acid sites although its surface properties can be altered by doping with metal and metal oxides.12Based on these characteristics, metal oxides have been studied in various ethanol based reactions. Zaki studied Fe2O3 and Mn2O3, among other oxides, in the ethanol dehydration reaction.13 Values of surface acidity, estimated by adsorption of pyridine (stronger acid sites) and piperidine (overall acidity), were intimately linked with the catalytic activity towards the dehydration products, ethylene and diethyl ether. In this referenced study, Fe2O3 and catalysts containing this metal oxide exhibited better behavior in comparison with Mn2O3 owing to its higher degree of strong acid sites. On the other hand, Mn2O3 has also been found suitable as an oxidative dehydrogenation catalyst, thanks to the presence of basic sites introduced by doping the oxide's framework with transition metals such as Co2+, obtaining over 80% selectivity towards acetaldehyde.14 As for the proper basic oxides such as ZnO, MgO, hidroxyapatites and their mixtures, these preferentially lead to the dehydrogenation products.15,16 Nevertheless, although basic sites are needed, it has been established that the selectivity towards acetaldehyde and further condensation products require the presence of acid–basic pairs.17 In this referenced publication, Di Cosimo et al. state that the O2− anion of pure MgO is responsible for its strong basic sites and, although this basic property is necessary for the dehydrogenation pathway, an initial step involving ethoxide formation requires Lewis acid–strong base pairs. These authors achieved their goal of designing successful dehydrogenation catalysts, maintaining the strong basic sites of MgO and introducing the Lewis acid sites, by incorporation of Al in the optimal amount. Other authors have also obtained high dehydrogenation activities combining basic properties of one oxide with electronic or redox properties of another. Such is the case of noble metal free ZnO–CeO2 catalyst, where the optimum proportion of Zn/Ce, on one hand, offers the right population of basic sites while, on the other hand, the Ce4+ to Ce3+ reduction is accompanied by the formation of oxygen vacancies, and as a whole, this catalysts yields higher activities.16
Regarding the ethanol transformation reaction, apart from the product orientation aroused by the acidity or basicity of the transition metal oxide, the incorporation of metallic nanoparticles can also tune both activity and selectivity towards the desired products. Hence, the choice of the most adequate metal active site is very much appreciated. In this sense, various publications involve the use of supported noble metals. Idriss summarizes the results obtained in the study of ethanol reactions employing noble metals such as Pt, Pd, Rh and Au supported on ceria in a review.18 In his work, the difference in selectivity towards one or the other products is directly related to the characteristics of the active phase. For example, the addition, of the metals (in general) on ceria inhibits its selectivity towards the formation of acetates (oxidation of ethanol) whereas the dehydrogenation reaction towards acetaldehyde and its corresponding higher condensation products is enhanced by the introduction of Pt or Pd. Of all these noble metals, Rh exerted a singular behavior as it catalyzes the breaking of C–C bond. Actually, this accused feature to readily dissociate C–C bonds, makes Rh a suitable candidate for a variety of reactions involving ethanol such as steam reforming,19 oxidation,20 etc. since this C–C cleavage is necessary prior to the production of CO2. In other works as that published by Gonçalves-Alonso et al., influence of the addition of noble metals, Pd and Ru, on Cu/Nb2O5 were studied in the ethanol reforming reaction.21 In this reaction it is known that hydrogen formation occurs mainly via dehydrogenation reaction where acetaldehyde formation is the first step involved. The addition of Ru and Pd not only enhance the activity and selectivity towards the dehydrogenation route, respectively, but also decreased the deactivation behavior of the un-modified Cu/Nb2O5 catalyst.
Among the noble metals, gold is becoming a first choice in several reactions owing to its extraordinarily high activity under relatively mild conditions.22 Its efficiency in catalysis can be questioned owing to the fact that it is the most noble metal known, however on the contrary it has been proven otherwise. An example is in the acetylene hydrodechlorination reaction.23 In this publication, gold is reported to have superior behavior not in activity but more importantly because it is exclusively selective to the desired product, vinyl chloride monomer, while the appearance of byproducts is suppressed. Various studies focus on the unique catalytic properties of gold. An important parameter to be highlighted is the particle size of Au nanoparticles employed. It is known that its singularity lies in the unsaturated surface atoms which would be enhanced when coordinated in small clusters. In fact, it is a proven point that particles must not be larger than 5 nm.24,25 For this reason, various studies focus on new synthesis methods or routes to produce small and well dispersed Au nanoparticles. This can be achieved by encapsulation in zeolites,26 thiolate-protection of metal nanoclusters,27 incorporation of additives,28 etc. In this latter reference, additives such as Ce and La adjust the structural and electronic properties of the support and hence affect Au nanoparticle size distribution. Other approaches such as speciation-controlled incipient wetness impregnation (ScIWI) take into account calculated thermodynamic species distribution diagrams as well as synthesis controlling parameters, such as pH, producing Au nanoparticles as small as 1 nm.29
The right combination of metal active phase and support is crucial to the catalytic behavior of the catalyst. Titania supported gold catalysts exert catalytic activity under milder conditions than with supports such as alumina or silica.30,31 Very recently, J. Quesada et al. analyzed the role of Au nanoparticles supported on TiO2 in the ethanol condensation reaction obtaining improved results, when compared to the bare support, in particular as refers to dehydrogenation activities.32 As was previously mentioned, when employed without incorporation of metal phase, metal oxides have also shown catalytic activity. For the partial oxidation of ethanol to acetaldehyde, TiO2 gave highest activity results at lowest reaction temperature when compared with Al2O3, SBA-15 (mesoporous SiO2) or hydrotalcite.33 On other oxides such as γ-Al2O3, the Lewis acid sites are said to be responsible for the dehydration of ethanol where ethylene is formed through the unimolecular pathway or diethyl ether through the bimolecular route.34 A. Gazsi et al.6 carried out a comparative study of ethanol adsorption and decomposition employing catalysts based on Au supported on various materials (Al2O3, CeO2, TiO2, SiO2, MgO, and Norit) of different acid–base and redox properties. Results obtained in their work gave insights to the role of Au as well as the influence of the different supports employed. In general, supported Au nanoparticles were active for the catalytic dehydrogenation of ethanol. On one hand, Au was said to enhance the formation of the ethoxy species, monitored by TPD and DRIFTS experiments, especially when supported on SiO2. On the other hand, Au/Al2O3, gave highest selectivity towards dehydration products. As far as the deactivation of catalysts is concerned, CeO2 supported Au catalyst presented the best behavior. The lack of deactivation in this case was said to be due to the interface between Au and the partially reduced support.
In this contribution, the results obtained in the catalytic transformation reaction of ethanol over Au NPs supported on ZnO tetrapods and on a commercial TiO2 are presented. Both supports were chosen owing to their different unique properties in terms of acidity, reducibility, structure, morphology, etc. which would definitely exert different influence on the resulting catalysts. We present a synthetic route for the design of high-performance Au catalyst by deposition–precipitation. A comparative study is carried out between both series of supported catalysts employing different characterization techniques. The synthesis, structure–property correlation, and the relative activity of the systems are presented, focusing on the influence of the size of gold nanoparticles and their interaction with the support on the catalytic activity.
Experimental procedure
Preparation of supported gold catalysts
Commercial TiO2 (P25) supplied by Sigma Aldrich (BET surface area 35–65 m2 g−1) and ZnO tetrapods (henceforth denoted as ZnO) were employed as supports. Synthesis of ZnO tetrapods (12–28 m2 g−1) were carried out by chemical vapor deposition as reported elsewhere.35 Deposition–precipitation method was used in order to synthesize the gold catalysts. A solution of HAuCl4·3H2O (purchased from Sigma Aldrich) in a mixture water/methanol (15/1) was added to the selected metal oxide support. After ultrasonic treatment at 283 K for 5 minutes, it was treated with an aqueous solution of NaOH (0.2 M) until pH = 11. Then, the precipitate was stirred in an iced bath to obtain small nanoparticles (defined as -a), at room temperature to produce medium sized particles (defined as -b) or at 313 K to achieve particles of larger dimensions (defined as -c). This procedure lasted 5 h and thereafter the resulting samples were collected by filtration. Finally, the precipitate was washed with deionized hot water, until no Cl− was detected. The sample was dried overnight at 373 K in air. The corresponding catalysts were labelled AuTiO2-a, AuTiO2-b, AuTiO2-c, AuZnO-a, AuZnO-b and AuZnO-c. These samples were subjected to a pretreatment in flowing N2 at 573 K for 1 h previous to the dehydrogenation reaction tests.Instrumentation and measurements
All the samples (both catalysts and supports) were characterized prior to and after reaction by X-ray photoelectron spectroscopy (XPS), X-ray diffraction (XRD), diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS), transmission electron microscopy (TEM) and thermogravimetric analysis coupled to mass spectrometry (TGA-MS).XPS analysis was performed with an ESCA-PROBE P (Omicron) spectrometer by using nonmonochromatized Mg-Kα radiation (1253.6 eV). TEM micrographs were obtained from a JEOL JEM-2100F microscope at 200 kV. The mean diameter of the Au particles was calculated, based on a minimum of 100 particles, using the following formula: 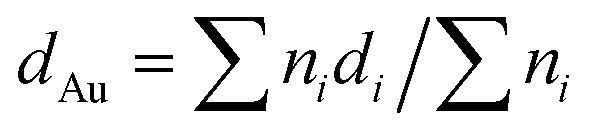 , being ni the number of particles with a diameter of di. Infrared spectra were obtained at room temperature on a FT-IR (Varian 670) spectrometer. XRD patterns were obtained on a Polycristal X'Pert Pro PANalytical diffractometer with Ni-filtered Cu/Kα radiation (λ = 0.1544 nm) operating at 45 kV and 40 mA. For each sample, Bragg's angles between 4° and 90° were scanned at a rate of 0.04 deg s−1. Thermogravimetric analyses were carried out in an SDT Q 600 apparatus on used catalysts under inert atmosphere (He 100 mL min−1) and in order to study evolution of desorbed products the TGA equipment was coupled with a quadruple mass spectrometer (Pfeiffer Vacuum Omnistar™ GSD 301). For this purpose, the following mass fragments were recorded taking into account the corresponding literature:36 H2 (m/z = 2), CO (m/z = 28 and 16), CO2 (m/z = 44), H2O (m/z = 18 and 17), ethanol (m/z = 31, 29, 45 and 27), acetaldehyde (m/z = 29, 44, 43 and 15), diethyl ether (m/z = 31, 59, 74 and 45), ethylene (m/z = 28, 27 and 26), butene (m/z = 41 and 56), crotonaldehyde (m/z = 41, 39, 70 and 69), butanal (m/z = 44, 43, 72, 41, 27 and 29), ethyl acetate (m/z = 43), 2-butenol (m/z = 57 and 29), benzene (m/z = 78 and 77), butanone (m/z = 43 and 72) and furan (m/z = 68 and 39). In cases where the most intense m/z signal was coincident in more than one specie, a secondary less intense signal had to be taken into account. The Au loading was determined employing inductively coupled plasma mass spectrometry (ICP-MS).
, being ni the number of particles with a diameter of di. Infrared spectra were obtained at room temperature on a FT-IR (Varian 670) spectrometer. XRD patterns were obtained on a Polycristal X'Pert Pro PANalytical diffractometer with Ni-filtered Cu/Kα radiation (λ = 0.1544 nm) operating at 45 kV and 40 mA. For each sample, Bragg's angles between 4° and 90° were scanned at a rate of 0.04 deg s−1. Thermogravimetric analyses were carried out in an SDT Q 600 apparatus on used catalysts under inert atmosphere (He 100 mL min−1) and in order to study evolution of desorbed products the TGA equipment was coupled with a quadruple mass spectrometer (Pfeiffer Vacuum Omnistar™ GSD 301). For this purpose, the following mass fragments were recorded taking into account the corresponding literature:36 H2 (m/z = 2), CO (m/z = 28 and 16), CO2 (m/z = 44), H2O (m/z = 18 and 17), ethanol (m/z = 31, 29, 45 and 27), acetaldehyde (m/z = 29, 44, 43 and 15), diethyl ether (m/z = 31, 59, 74 and 45), ethylene (m/z = 28, 27 and 26), butene (m/z = 41 and 56), crotonaldehyde (m/z = 41, 39, 70 and 69), butanal (m/z = 44, 43, 72, 41, 27 and 29), ethyl acetate (m/z = 43), 2-butenol (m/z = 57 and 29), benzene (m/z = 78 and 77), butanone (m/z = 43 and 72) and furan (m/z = 68 and 39). In cases where the most intense m/z signal was coincident in more than one specie, a secondary less intense signal had to be taken into account. The Au loading was determined employing inductively coupled plasma mass spectrometry (ICP-MS).
Catalytic test
Catalytic conversion of ethanol reactions were carried out at atmospheric pressure. In each test, 50 mg of catalyst diluted in glass beads were loaded in a glass tube (9 mm id) that served as a fixed-bed, continuous flow reactor. 15 μL min−1 of ethanol (99.8% purity) were diluted in helium (total flow rate of 20 mL min−1) and passed through the catalytic bed. At regular time intervals the reaction products were analyzed in a gas chromatograph (GC) apparatus, equipped with both TCD and FID detectors. This analysis system is directly coupled to the reactor exit. The identification of reaction products was achieved bearing in mind the retention times of pure reference standards. When present, peaks corresponding to unknown condensation products (trace amounts) were integrated using the response factor of butanol. The transformation reaction of ethanol was studied at 573 K during 400 minutes. Conversion (XETOH) and selectivity (S) were estimated employing the following equations:where [CETOH]in and [CETOH]out represent the total ethanol concentration in the inlet and outlet gas, respectively.
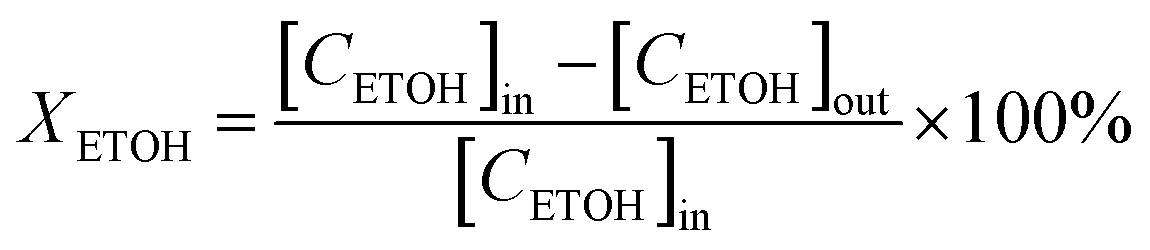
where ni and Ci are the number of carbon atoms and concentration of the product (i), respectively.
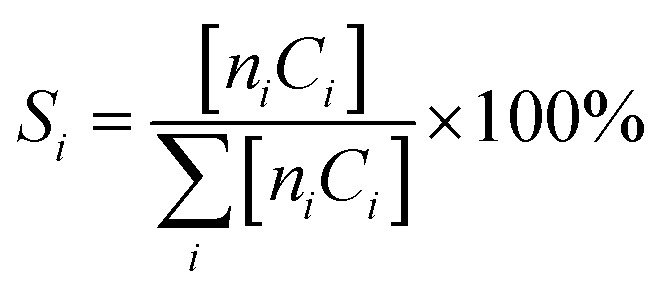
TOF was also calculated considering the number of moles of ethanol converted per surface atom per second. For this, the number of active sites at the surface of the catalyst was deduced from XPS analysis as has been done in other publications.37

Results
Catalysts characteristics prior to the reaction
Information on the degree of crystallinity and phase were obtained from XRD diffractograms. Fig. S1 (view ESI†) exhibits patterns of support as well as resulting catalysts. Peaks observed in the TiO2 support material belong to rutile and anatase phases in the relative ratio of 76%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :24% which is similar to literature findings for this type of commercial supports where ratios of 75%:25% are generally observed.38 On the other hand, the contribution of other phases such as brookite phase was not observed. The incorporation of Au nanoparticles made no apparent changes in the XRD pattern of the resulting catalysts when compared with that of the TiO2 support.
:24% which is similar to literature findings for this type of commercial supports where ratios of 75%:25% are generally observed.38 On the other hand, the contribution of other phases such as brookite phase was not observed. The incorporation of Au nanoparticles made no apparent changes in the XRD pattern of the resulting catalysts when compared with that of the TiO2 support.
As for the ZnO series all Au/ZnO catalysts displayed peaks of ZnO wurtzite phase (JCPDS-36-1451).39 Apart from these peaks owing to the support, in certain samples, peaks around 38.3, 44.5 and 64.5° were also visible owing to the diffraction planes (111), (200) and (220), respectively, of Au nanoparticles (JCPDS-04-0784).40 This is particularly visible in sample AuZnO-c (Fig. 1) while the other two samples displayed the same XRD patterns as the initial ZnO support. Hence, the former has larger particle sizes.
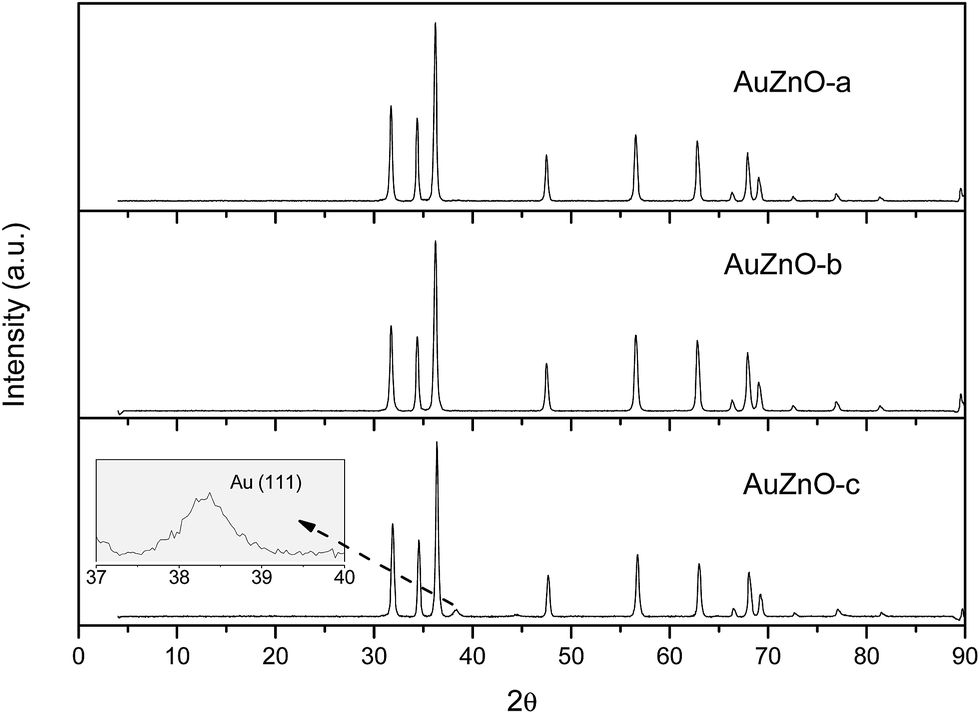 | ||
| Fig. 1 XRD diffractograms of AuZnO catalysts. | ||
TEM images as well as size distribution histograms of the TiO2 series are depicted in Fig. 2. When employing this support, sizes of 2.2, 3.4 and 5.2 nm were obtained as a consequence of the different temperatures employed in each case during catalyst synthesis. These particles are too small to be detected by XRD as was corroborated by the absence of Au (111) peaks at about 38°. As refers to the particle size distribution, AuTiO2-a exhibits a narrower distribution in comparison with AuTiO2-b and especially with AuTiO2-c where sizes of up to 11 nm are observed.
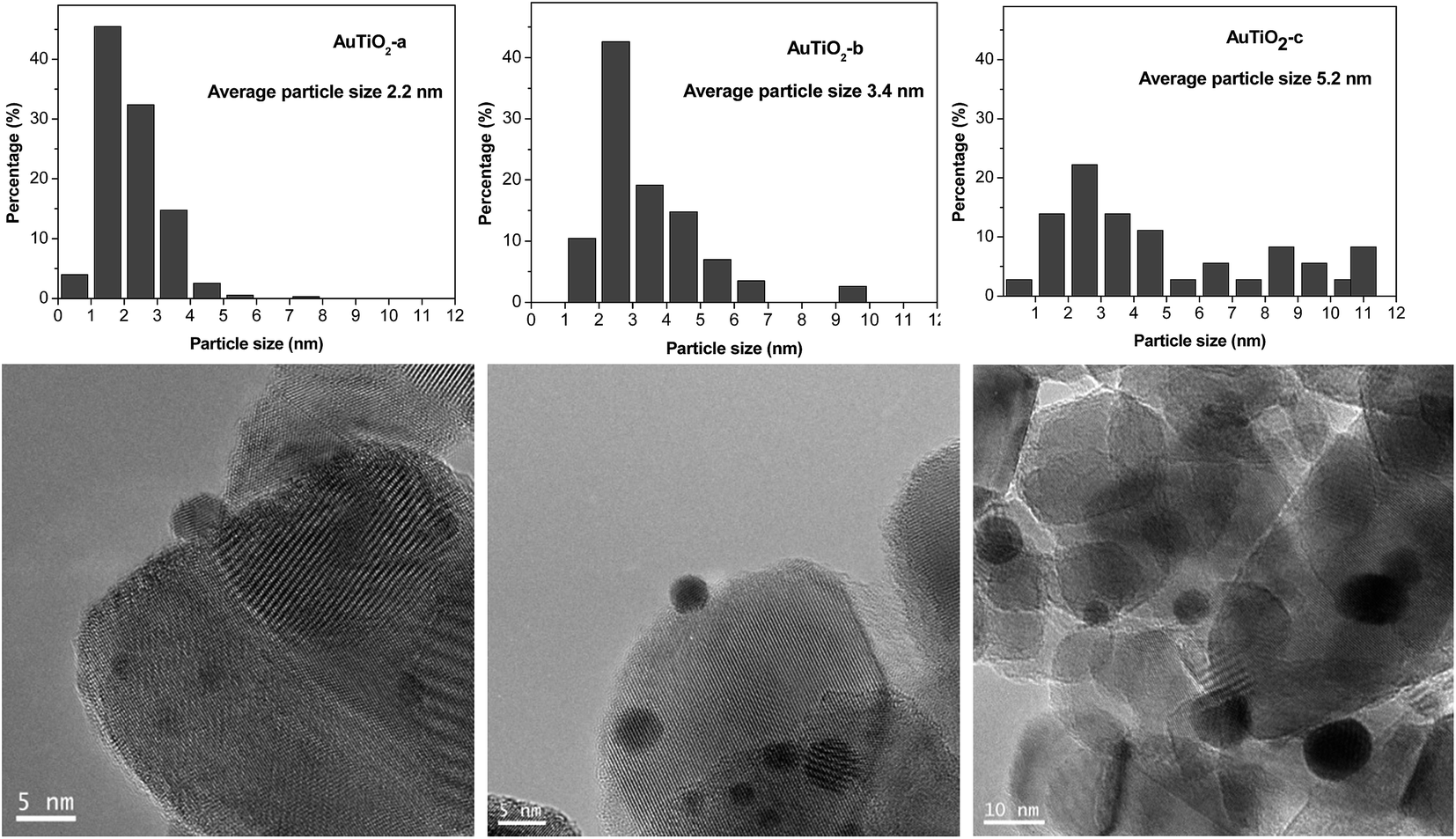 | ||
| Fig. 2 TEM micrographs and particle size histograms of TiO2 supported Au catalysts. | ||
Fig. 3 displays the TEM results for the ZnO series and in agreement with XRD patterns the following order of particle sizes was found: AuZnO-a < AuZnO-b < AuZnO-c. One can easily deduce that this support favors the formation of larger nanoparticles with respect to the TiO2 material. The presence of some tetrapods of ZnO formed from nanorods is clearly visible (insert in Fig. 3 AuZnO-c). These structures have a diameter ranging from 8–40 nm and lengths of 100 nm to 1 mm. In this series a wider distribution of Au NPs sizes is observed in comparison with the TiO2 series.
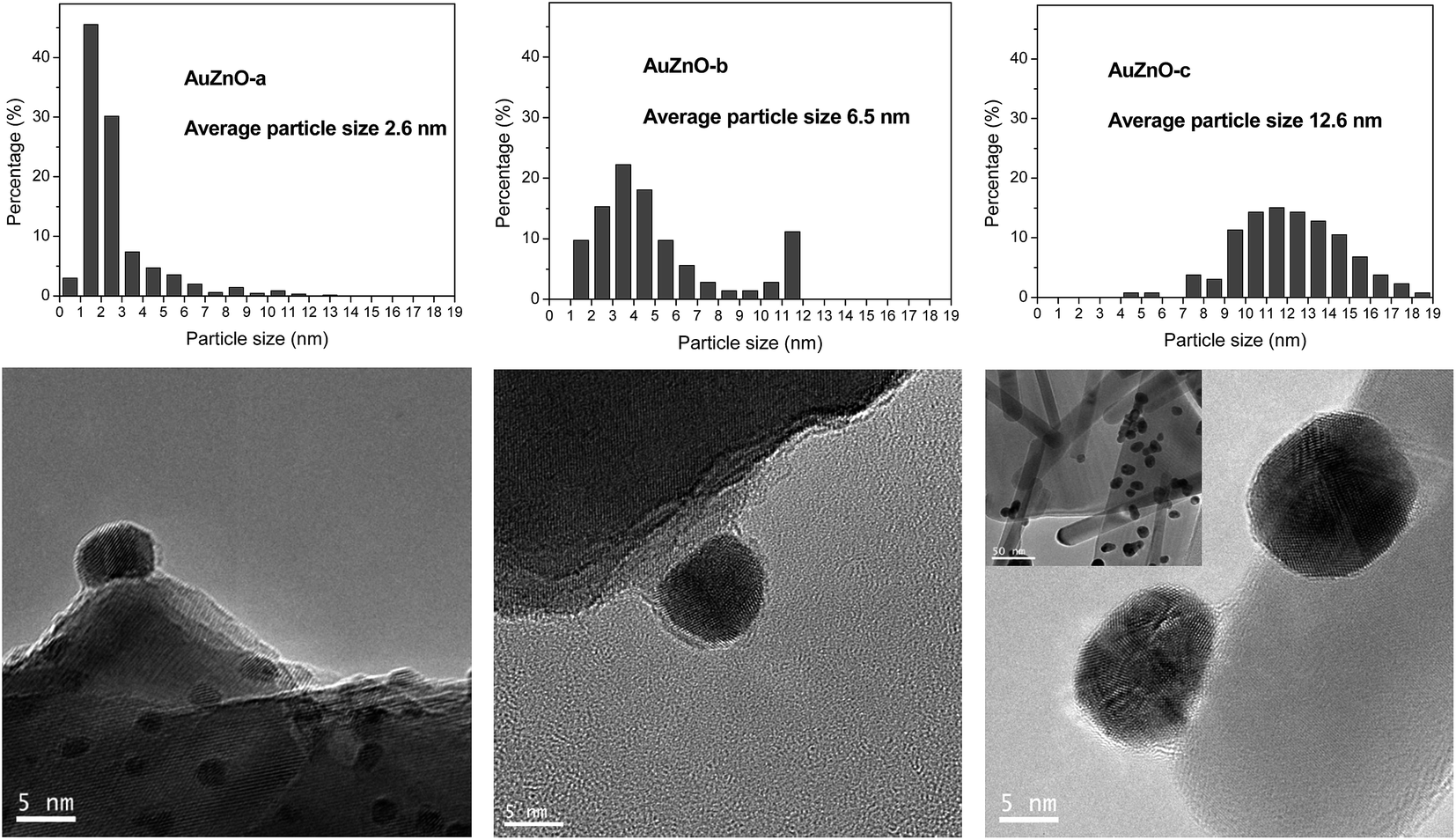 | ||
| Fig. 3 TEM images and particle size histogram of AuZnO-a, AuZnO-b and AuZnO-c. | ||
XPS analyses gave further insights as refers to surface composition of samples. As was expected, the gold surface composition detected in the fresh catalysts is in line with the degree of dispersion observed in TEM images, i.e., smaller particles give rise to higher surface percentage revealed by XPS. When expressed in terms of wt%. values range from above 7% for AuTiO2-a to below 2% for AuTiO2-c also hinting on the variation of degree of dispersion for all samples of similar metal loading of around 2% as was determined by ICP-MS measurements. As refers to the binding energies, the envelope of the Au 4f7/2 peak had a maximum at about 83.4 eV which is a bit lower than that normally observed for Au (0) implying the possibility of interaction between support and nanoparticles. At the same time, apart from the presence of metallic gold, there is a smaller contribution at about 84.9 eV (17% atomic) owing to the presence of Au(I). Similar results were obtained by Ousmane et al. where 15% of Au(I) was observed.41 It was said that in these catalysts composed of Au supported on titania, part of the Au(I) component would likely diffuse at the interface with the TiO2 support, thereby maintaining its oxidation state even after reaction. The deconvolution of the envelope into the two components of one of the AuTiO2 catalysts can be viewed in Fig. 4(a).
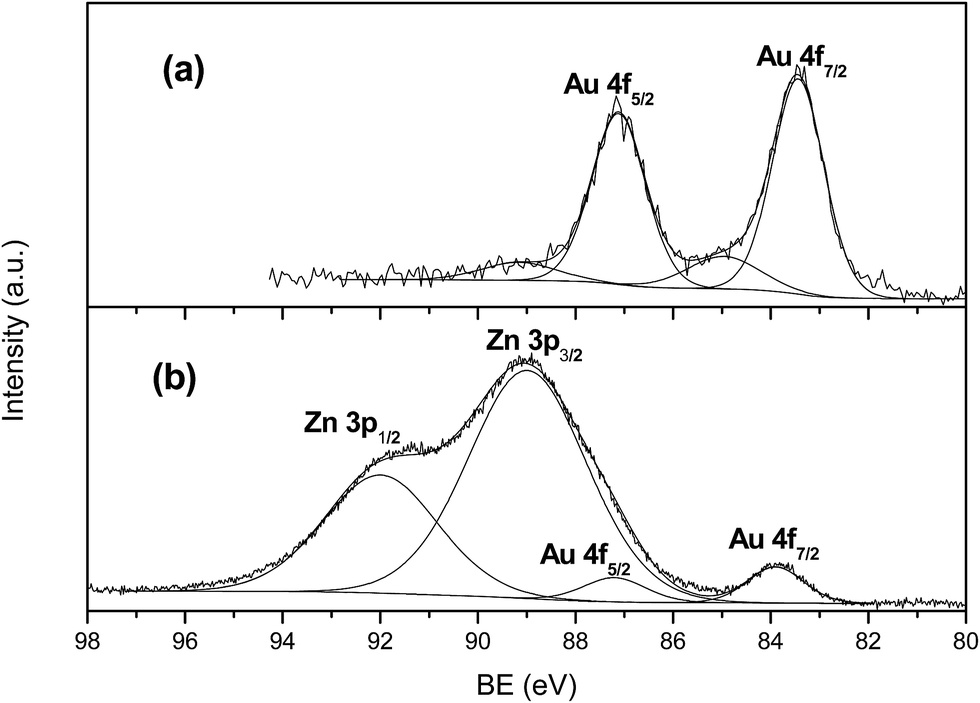 | ||
| Fig. 4 XPS spectra of Au 4f for (a) AuTiO2-a and (b) AuZnO-a. | ||
In the case of ZnO catalysts, surface atomic composition of Au expressed as wt% is about 4.5% (for samples of smallest Au NPs size) of the total catalysts mass which is higher than that obtained by ICP-MS (∼2%) owing to the small particles deposited at the surface of the metal oxide and hence its high dispersion. On the other hand, for these series of catalyst, Au 4f7/2 peak appeared close to 84 eV (Fig. 4(b)). This value is typical of metallic gold and is in agreement with studies employing samples of similar characteristics.40
Catalytic test
The catalytic transformation of ethanol carried out at 573 K during 400 minutes was studied with both series of catalysts. In all cases activity, selectivity towards reaction products and stability of catalysts were evaluated. Blank experiments involving supports without Au nanoparticles were also conducted. In Fig. 5 conversion versus reaction time is represented for the two series. Commercial TiO2 material exhibited a rather low activity. Conversion values were below 3% during all 400 minutes of reaction. The bare ZnO support was also tested and conversion was equally low (about 4%) at the temperature studied. As for the titania-supported Au catalyst, conversion values were significantly higher in all catalysts of this series than that of the bare support. Scalbert et al.15 studied the possible reaction routes of transformation of ethanol to butanol and stated that the presence of metal, in bi-functional solids, reduced the reaction temperature when compared with metal free catalyst. A main difference is in the hydrogenation step where hydrogen atoms are involved in the former while for the latter, it is more likely to occur via hydrogen transfer between two molecular species.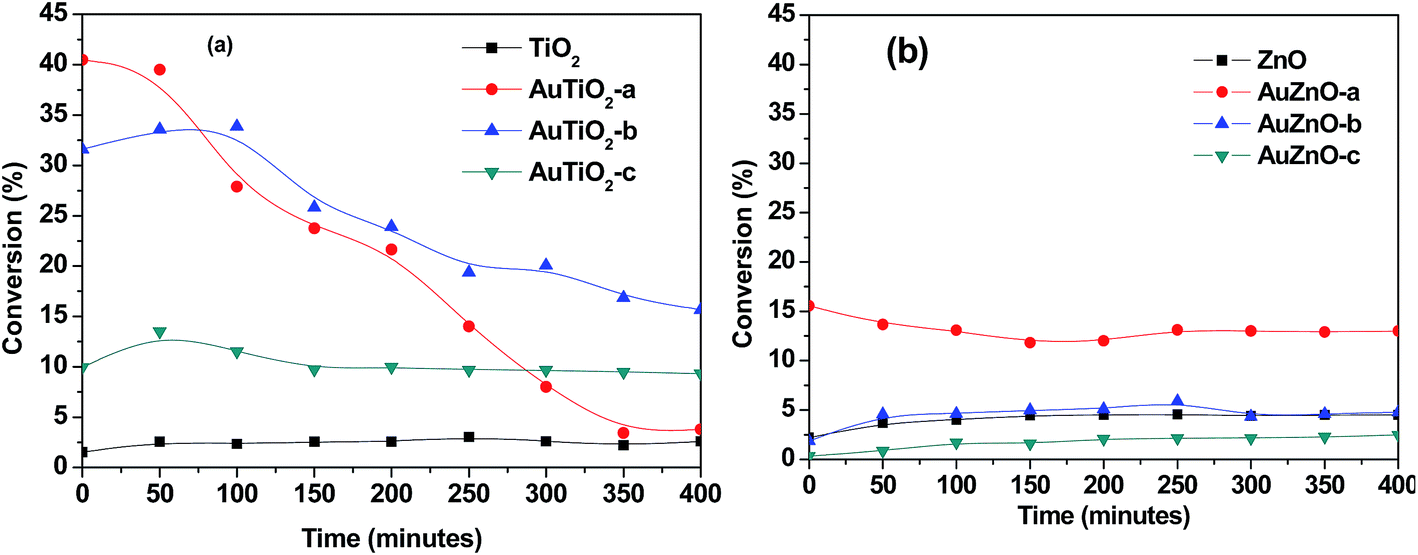 | ||
| Fig. 5 Conversion versus time for studied catalysts (T = 573 K). | ||
Initial conversion in this work refers to values after 5 minutes on stream. These initial conversion values follow the order: AuTiO2-a > AuTiO2-b > AuTiO2-c, and keeping in mind the particle size estimated by TEM, as was mentioned above, these results suggest that activity is inversely proportional to particle size, i.e., smaller nanoparticles yield higher conversion values, which is consistent with a higher exposed surface area of active metallic sites. Surprisingly enough, some authors highlighted that the optimal catalytic activity of Au/SiO2 catalysts was found for Au nanoparticles of about 6 nm which exhibited higher conversion in the ethanol dehydrogenation reaction than the smaller or larger ones.5 Also Zheng and Stucky found higher conversions in the ethanol oxidation for the medium particle size of 6.3 nm, in comparison with the larger (8.2 nm) or the smaller (3.5 nm) Au nanoparticles supported on SiO2.42 However, in our work, TiO2 supported Au nanoparticles of small size (either AuTiO2-a or AuTiO2-b) display significantly higher conversion in comparison with the 5.2 nm ones (AuTiO2-c). On the other hand, catalyst deactivation follows the opposite direction. The catalyst with smallest Au nanoparticles, AuTiO2-a, rapidly deactivates (∼90% deactivation) while that of largest particles, AuTiO2-c, maintains its conversion practically constant (∼7% deactivation). As for AuTiO2-b, the obtained conversion slightly increases during the first 100 min on stream and then it stars to decrease with a rate which is intermediate between the two extremes (∼50% deactivation). In other words, the smaller the Au particle size, the higher its deactivation rate, as a consequence of the higher conversion levels achieved from early stages of the reaction. This results in a change in conversion values at the end of the 400 minutes duration of experiments, as can be viewed from Fig. 5, where conversion has an new order: AuTiO2-b > AuTiO2-c > AuTiO2-a. It is worth to note that carbon balances decrease until values close to 70% in this series of catalysts which suggests the possible retention of reaction products at the catalysts surface, as will be discussed later.
As for the catalytic behavior of the ZnO series, two main differences can be highlighted when compared with the TiO2 series. In the first place, stability over 400 minutes of reaction is much higher as can be seen in Fig. 5 where deactivation is much lower in (b) than in (a). In second place, initial conversion values are considerably lower for the ZnO series (Fig. 5(b)). In this case, owing to their lower activity and taking into account the larger particle sizes obtained in general, in some cases the conversion of the supported catalyst is lower than that of the support on its own. It is known that for Au to act as good catalysts particle sizes have to be below 5 nm.24 Therefore it could be fully understood why AuZnO-c (12.6 nm) has lower conversion values than ZnO. Here, AuZnO-c is not only inactive but also its presence reduces the exposed surface of the support thereby reducing its reactivity.
When comparing two samples of similar Au nanoparticle size and metal loading of ∼2% (AuTiO2-a and AuZnO-a) the differences in conversion levels are quite obvious. While AuTiO2-a (2.2 nm) offers initial values of 40%, thereafter diminishing to ∼5%, AuZnO-a (2.6 nm) with initial conversion value of 15.6% reduces until 13%. The differences between both catalysts can be attributed to the influence of the support on the catalytic activity. On one hand, and generally speaking, TiO2 favors the formation of smaller gold nanoparticles with respect to ZnO. On the other hand, catalysts of similar particle sizes belonging to the two series exhibit different catalytic behavior.
Initial TOF values were estimated for all catalysts with conversions above those of their corresponding supports and of particle sizes ≤ 5 nm. Therefore, AuZnO-b and AuZnO-c were discarded as their activity is below or almost the same as that of ZnO and nanoparticles were above 5 nm. For this same reason and as has been stated earlier, the complete characterization study of these two catalysts was not conducted or included in the corresponding section. The estimated values are given in Table 1, where the following order was observed: AuTiO2-b > AuTiO2-a > AuTiO2-c > AuZnO-a. AuTiO2-b of medium particle size, 3.4 nm, is the catalysts that offered best results doubling that of the AuTiO2-a which presented higher initial conversion values whereas this latter of smaller particle size (2.2 nm) tends to deactivate more rapidly. Of all four catalysts, that supported on ZnO offers the lowest TOF value and the formation of successive condensation products is greatly reduced.
| Sample | Particle size nm (TEM) | TOF (s−1) |
|---|---|---|
| AuTiO2-a | 2.2 | 0.088 |
| AuTiO2-b | 3.4 | 0.164 |
| AuTiO2-c | 5.2 | 0.084 |
| AuZnO-a | 2.6 | 0.054 |
| AuZnO-b | 6.5 | — |
| AuZnO-c | 12.6 | — |
Product distribution was also studied. Selectivity at early stages of the reaction as well as after 400 minutes on stream is depicted in Fig. 6. The main product obtained with the bare TiO2 support was the acid-catalyzed dehydration product diethyl ether, followed by acetaldehyde, ethylene and butene. This should be expected as TiO2 is said to be an amphoteric oxide exhibiting medium and strong Lewis acid sites, weak Brønsted acid sites and a few weakly basic sites.11 Significant changes are observed with the introduction of Au in the catalysts. In general terms, acetaldehyde is the main product while the formation of diethyl ether has been greatly reduced agreeing with literature findings.43 This is most significant when employing catalysts of smaller Au nanoparticles, where values of about 70% (acetaldehyde selectivity) are observed. As can be seen in Fig. 6, for catalysts with smaller Au sizes, crotonaldehyde, originated through the base-catalyzed aldol condensation mechanism from acetaldehyde, is the second most important product after this latter although its value reduces drastically over the 400 minutes reaction period. Other secondary products detected (C4 alcohols and butanal) are also involved in the Guerbet reaction mechanism from ethanol.44 As was mentioned earlier, samples with smallest Au particle sizes (AuTiO2-a) experience severe deactivation behavior. Hence, at lower conversion values, subsequent condensation products are not likely to form. As for samples that exhibited lower initial activity and greater stability, product distribution is less distorted.
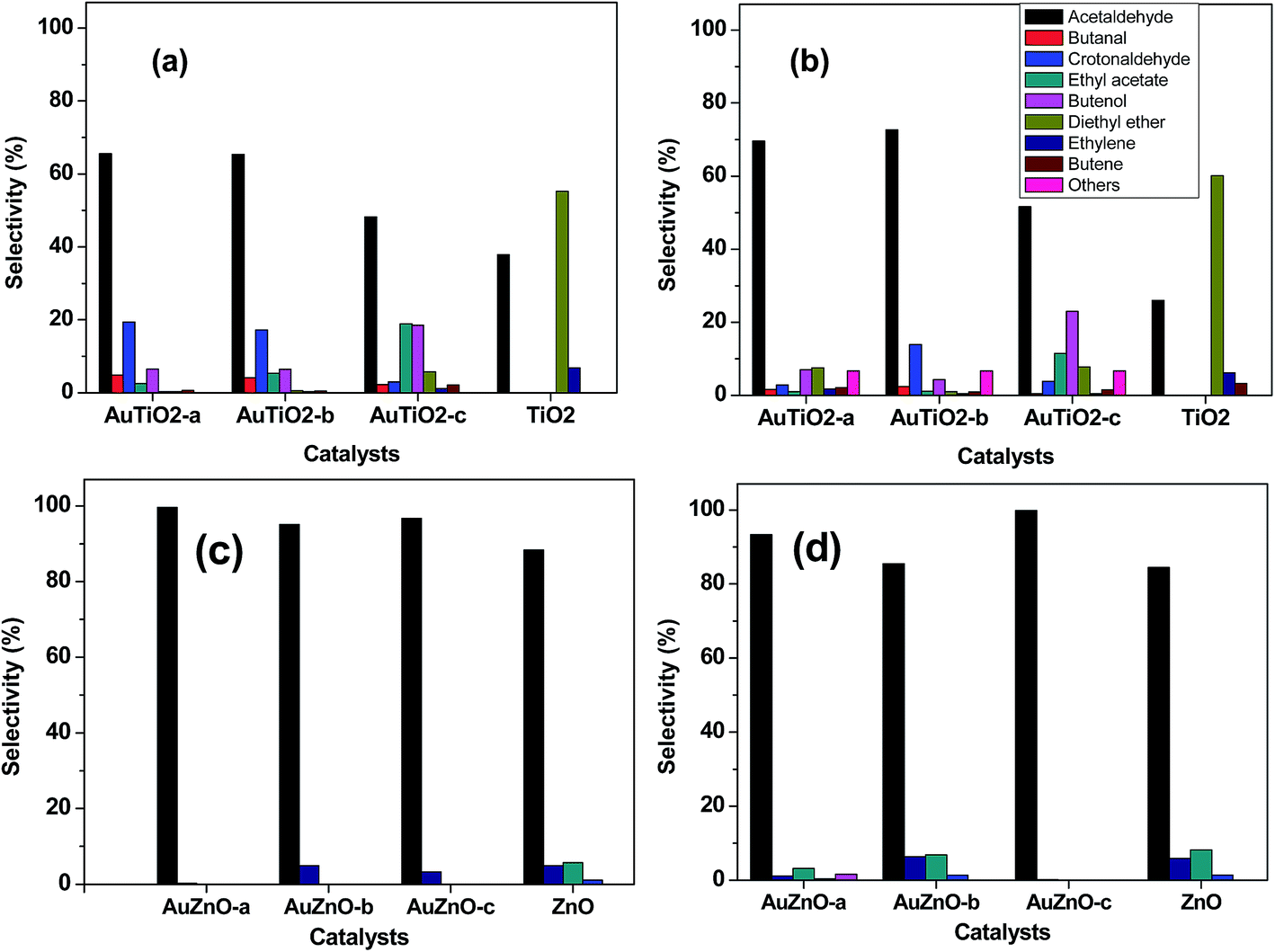 | ||
| Fig. 6 Product distribution at initial ((a) and (c)) and after 400 minutes ((b) and (d)) of reaction for AuTiO2 series and AuZnO series (T = 573 K). | ||
As for the ZnO series, acetaldehyde is by far the main product observed (above 80%) with the bare support and in contrast to TiO2 support the presence of diethyl ether, which was its primary product, was not detected. In our previous work, we studied a series of ZnO materials of different morphologies and surface functionalities.45 In all cases, the dehydrogenation product, acetaldehyde, was obtained with selectivity values of 84–94%. The formation of this primary product was said to involve the presence of acid–basic pairs on the surface of the catalyst. In our actual work, the ZnO support also favors the formation of acetaldehyde while subsequent condensation products such as crotonaldehyde and ethyl acetate (from the dimerization of acetaldehyde or direct dehydrogenation of ethanol46) are just about 1 and 5%, respectively. At the same time, ethylene, the dehydration product owing to the acidic properties of this material is present in about 5%. Very little variations are observed during the 400 minutes of reaction. It should be noted that at the end of the reaction, there is an increase in ethyl acetate at the expense of acetaldehyde which has reduced from 88 to 84%. For all three Au supported ZnO catalysts employed in the catalytic test, acetaldehyde was the main product with selectivity values of over 95% in all cases at the initial stages of the reaction. Hence, the incorporation of Au favored the dehydrogenation reaction pathway in comparison with the bare support. On the other hand, selectivity towards ethylene is below 5%. Evolution of main product, acetaldehyde, towards secondary products such as ethyl acetate, crotonaldehyde and 1-butanol (or 2-butenol) was observed during the 400 minutes of reaction studied at 573 K (Fig. 6(c and d)).
Catalysts characteristics after reaction
XRD measurements performed on catalysts after reaction were similar to those obtained with the fresh catalysts, indicating no apparent sintering of Au NPs under the reaction conditions, and for the sake of brevity the diffractograms are not included in this manuscript but in the ESI section (Fig. S1-c†).Infrared spectroscopy can reveal valuable information concerning the presence of surface carbonaceous species on deactivated catalysts.47,48 DRIFTS measurements were carried out before and after reaction and differences between both spectra were analyzed. Besides the studied catalysts, DRIFTS experiments were also performed on support materials. The spectrum of TiO2 is included in Fig. S2.† According to the literature,49 the broad band from about 3380 to 3400 cm−1 can be ascribed to the stretching vibration of water molecules. This is further confirmed by the presence of a peak at about 1600 cm−1 owing to the bending vibration of the O–H group. This latter contribution also corresponds to the presence of Ti–O groups of titania based samples which in addition exhibit bands around 800–1000 cm−1 due to Ti–O–Ti vibrations. Similar peaks were observed in the fresh Au–TiO2 catalysts.
As for the used catalysts, significant differences can be found when compared with their former spectra. Fig. 7 displays the spectrum of AuTiO2-a and AuZnO-a catalysts (prior to and after reaction), where the presence of coke can be identified by the typical bands of hydrocarbon molecules in the most important regions at 2850–3000 cm−1 and 1700–1300 cm−1. The rest spectra of AuTiO2 materials are depicted in Fig. S2 (ESI†). The bands in the region of 2850–3000 cm−1 correspond to aliphatic (asymmetric and antisymmetric stretching)50 and single-ring aromatics.47,48 Distinctive bands at 2958 and 2870 cm−1 of CH3 groups appeared, together with the characteristics bands of –CH2 and –CH groups at 2930, 2900 and 2855 cm−1. Also, distinctive bands at 1377 and 1443 cm−1 corresponding to CH3 symmetric and antisymmetric deformation,50,51 respectively, appeared. This suggests that carbonaceous species could consist of aliphatic hydrocarbons or unsaturated naphthenes.48
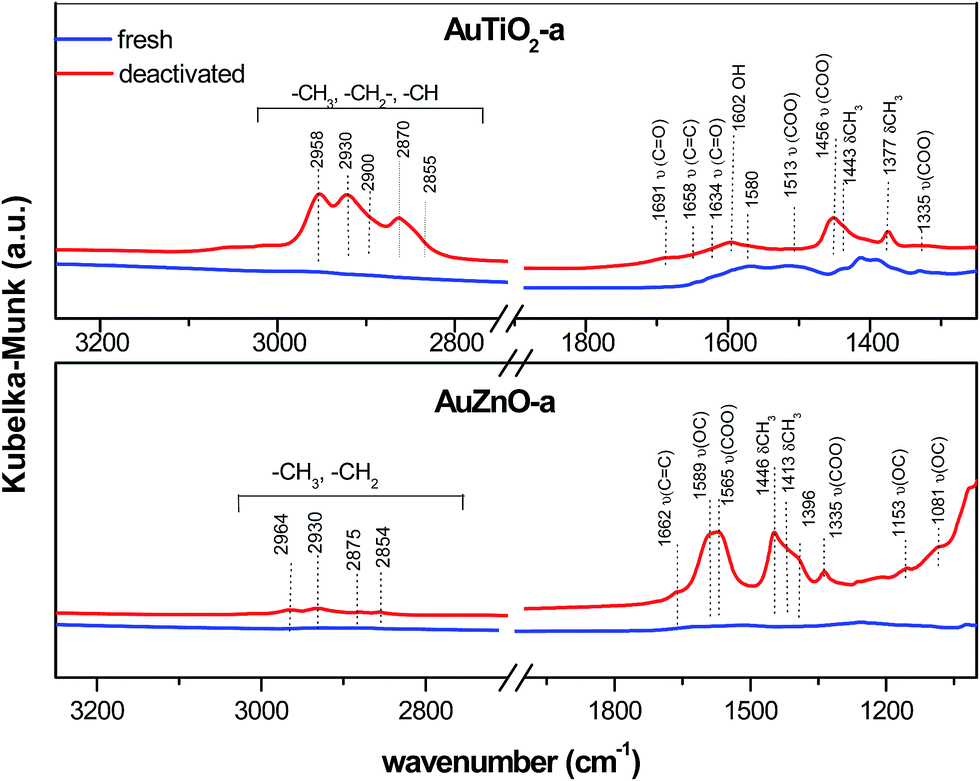 | ||
| Fig. 7 DRIFT spectra of fresh and used AuTiO2-a and AuZnO-a catalysts. | ||
Bands in the region 1700–1300 cm−1 include molecular vibrations that can be ascribed to different functional groups (Fig. 7). The presence of acetate species can be deduced by the 1456 and 1513 bands, due to –COO– symmetric and antisymmetric stretching, respectively.52 Spectral characteristic features of acetaldehyde at 1691 cm−1 ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) was also found in the used catalysts. Characteristic bands of the condensation product crotonaldehyde appeared at 1658 and 1634 cm−1.53 It is worth to note that the presence of these peaks was most pronounced in samples with smaller Au nanoparticles (Fig. S2†).
O) was also found in the used catalysts. Characteristic bands of the condensation product crotonaldehyde appeared at 1658 and 1634 cm−1.53 It is worth to note that the presence of these peaks was most pronounced in samples with smaller Au nanoparticles (Fig. S2†).
DRIFTS measurements were also performed on the ZnO based samples. For comparison reasons a spectra of a catalyst belonging to this series, AuZnO-a (prior to and after the reaction) is included in Fig. 7. Measurements of AuZnO-b and AuZnO-c were not included in Fig. S2† owing to the large particle sizes determined by TEM, which as was earlier discussed in the corresponding section, did not have relevant influence on the catalytic activity. As can be observed, differences detected in this studied series differ from that of TiO2 based samples. On one hand, bands in the 2980 to 2850 cm−1 range owing to νCH3 mode of aliphatic hydrocarbons54 have also increased in the used samples when compared with the corresponding fresh ones although in this case the increase is much smaller than was observed for the TiO2 series. Nevertheless, and in this set of samples, as can be seen in Fig. 7, this region includes four bands at about 2964, 2930, 2875 and 2854 cm−1, and together with those found at lower frequencies (1153 and 1081 cm−1), are said to correspond to ethoxy species adsorbed on ZnO supported catalysts.55 On the other hand, the relative ratio of bands owing to the presence of surface oxygenates (e.g. νCO (∼1589 cm−1)) to –CH groups (e.g. δCH3 (∼1446 cm−1)) for used samples is higher than in the case of TiO2 supported samples. Bands at about 1565 cm−1 could be ascribed to the acetate species obtained from the dehydrogenation of ethoxy species.55,56 No apparent changes between fresh and used samples are observed in the νOH band at about 3560–3670 cm−1 region which corresponds to the surface hydroxyl groups of ZnO (Fig. S2†).57
XPS analyses gave further insights as refers to surface composition of samples before and after reaction. Table 2 summarizes atomic percentages of Au before and after reaction. These XPS results can also be visualized in Fig. S3 (ESI†) where, as can be observed, C 1s significantly increased after reaction at the expense of the other components (Au, Ti and O). The ratio of Au to metal oxide support reduces for the catalyst of smallest particle size, AuTiO2-a, due to the carbon species which are deposited both on the gold and metal oxide support. Table 2 also includes the values of the ratio (used with respect to fresh) of atomic % of carbon content. From this data one can observe how catalysts with smaller particles retain higher carbon components. These results are in agreement with those obtained by DRIFTS.
| Sample | Fresh | Used | Increase in carbon deposits: used with respect to fresh (Cused/Cfresh) |
|---|---|---|---|
| Au (at %) | Au (at %) | ||
| AuTiO2-a | 0.82 | 0.2 | 2.80 |
| AuTiO2-b | 0.33 | 0.09 | 2.91 |
| AuTiO2-c | 0.19 | 0.20 | 1.31 |
| AuZnO-a | 0.49 | 0.42 | 1.10 |
As refers to XPS analyses, results for the AuZnO catalysts were quite different from those obtained for AuTiO2. A quick view at Table 1 and Fig. S3,† highlights these differences. In this case, surface atomic composition of Au doesn't seem to vary much while the increase in C atomic ratio at the surface of samples after reaction is insignificant.
To further confirm and identify the type of carbon deposits present on the surface of spent catalysts, desorption of adsorbed species was followed by TGA-MS on the used samples. For this, samples were heated until 1173 K at a rate of 5 K min−1 under a 100 mL min−1 flow of He (TGA) and the products evolved were followed by mass spectrometry (MS). Fig. 8(a) presents the weight loss experimented due to the desorption of the carbon products formed during the reaction while Fig. 8(b) shows the mass fragments desorbed during this heating process for the sample which presents most drastic changes after reaction, AuTiO2-a. Weight loss (∼3%) is observed at about 658 K as can be deduced from the thermogravimetric analysis profile. This temperature value is coincident and is within the maxima observed, between 630 and 672 K, of the desorbed species detected by mass spectrometry (Fig. 8(b)). As can be viewed, the mass fragments corresponding to the following carbon compounds were found: acetaldehyde, ethylene, butene, crotonaldehyde, ethyl acetate, 2-butenol, butanol, furan and benzene. In our catalytic tests we did not identify these latter two aromatic compounds, which suggests that they remain adsorbed at the catalyst surface. Nadeem et al.24 also detected these aromatic compounds when studying the reactions of ethanol by TPD and IR spectroscopy on the surface of AuTiO2 catalysts. They proposed the reaction mechanism formation of benzene over AuTiO2 catalysts as follows: adsorbed crotonaldehyde and acetaldehyde react yielding 2,4-hexadienal; thereafter, Au nanoparticles break the C–H bond of the methyl group which after intramolecular cyclisation followed by H2O elimination may give benzene.
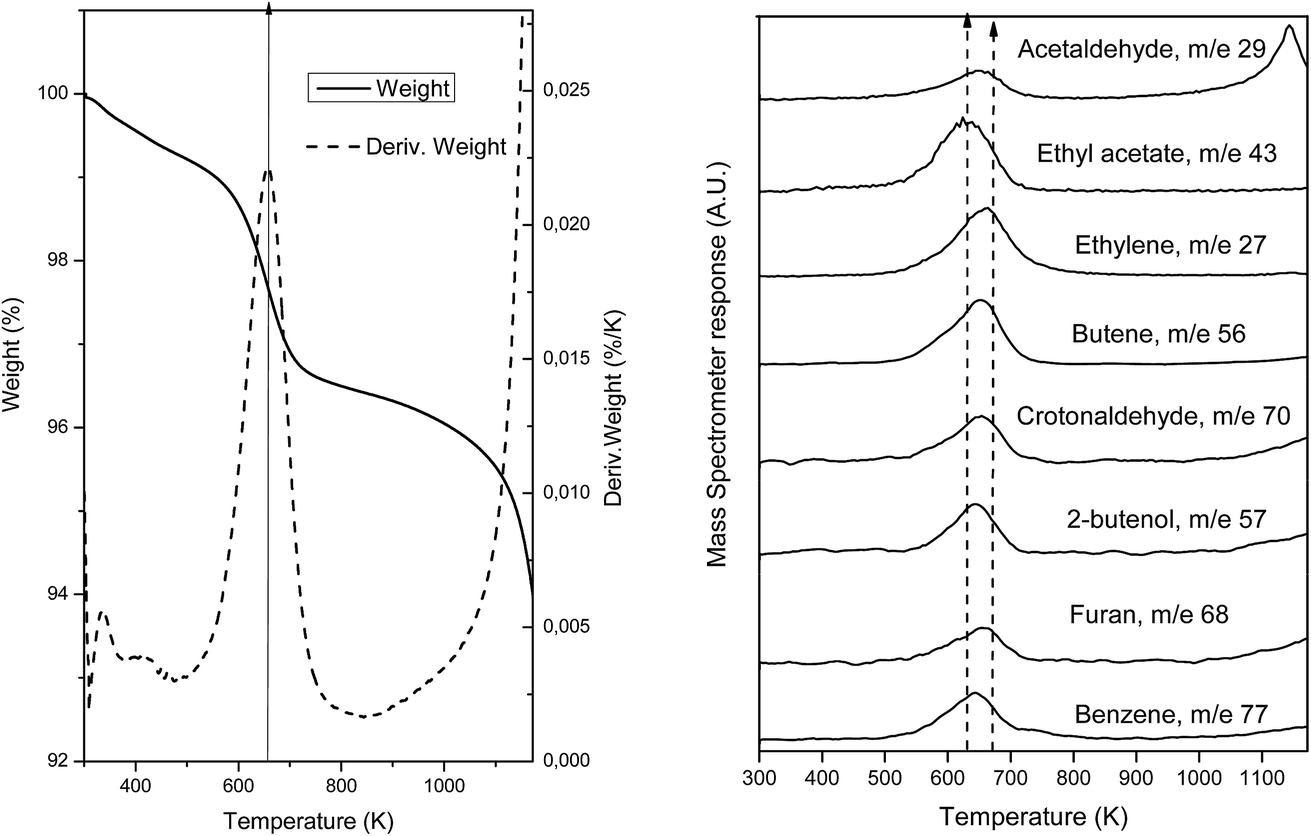 | ||
| Fig. 8 TGA-MS of AuTiO2-a showing desorbed species of used catalyst. | ||
In our study, MS results are not presented to scale, and so serve only for qualitative and not quantitative purposes. The existence of all these species confirms DRIFTS and XPS analyses results, proving that the presence of carbon products adsorbed on the surface reduces/blocks the area of active sites exposed and this could interfere with the catalytic activity as observed in the catalytic test section of this work.
Desorption study of carbonaceous compounds was also followed by this same TGA-MS analysis for the ZnO series and in this case, it is worth to highlight that the weight loss in this catalyst is less than 1% wt confirming the lower quantity of coke deposits in comparison with AuTiO2 catalysts (∼3.5% wt loss). Mass fragments corresponding to ethanol, acetaldehyde, ethyl acetate, ethylene and diethyl ether, were observed. This last listed product (diethyl ether) was not observed within the products analysed by GC during the catalytic test, but may be present in trace concentration or may have remained adsorbed at the catalyst surface, which would be coincident with the ethoxy species observed by DRIFTS. In general terms, these results differ from that obtained in the TiO2 series where heavier condensation products (furan and benzene), were retained at the catalysts surface. All this would be further highlighted in the following section.
Discussion
Our results indicate that the deactivation observed on the TiO2 series is due to the deposition of carbonaceous species formed during the reaction and strongly adhered to the surface of the catalyst which results in activity loss due to the blockage of active sites. XPS analyses results as well as DRIFTS corroborated this explanation. This was further confirmed by TGA-MS, where the type of deposits desorbed from the surface can be related to the selectivity towards the different products obtained during the catalytic test. Recalling from the previous section, for series TiO2, acetaldehyde, ethylene, butene, crotonaldehyde, ethyl acetate, 2-butenol, benzene, furan and butanol were observed whose presence on the surface reduces the area of active sites exposed thereby diminishing the catalytic activity and causing deactivation. As for the ZnO series, desorption of the following carbon deposits: ethanol, acetaldehyde, diethyl ether, ethyl acetate and ethylene, were observed. The absence of higher condensation products (butane, furan and benzene) in this series is evident and marks the difference between both series. At this point, one could state that, the mass fragments observed in this desorption study are in agreement with the selectivity mainly towards primary reaction products determined for the ZnO catalysts which don't seem to lead to high deactivation.Taking into consideration results obtained so far as refers to extent of reaction reached as confirmed by the catalytic test, products found at the catalyst surface as was revealed by TGA-MS experiments and IR bands observed in the DRIFTS experiments, Fig. 9 was designed. In this illustration where all these results are combined, one catalyst of each series (of similar particle size) is presented in order to detect differences. Various remarks can be made on viewing both graphs. On one hand, there are significant differences in terms of product distribution and IR bands between both graphs. On the other hand, these IR bands match perfectly with the products predicted in each case. Some of the bands identifying the products desorbed are marked highlighting the concurrence found on combining the characterization and catalytic tests results obtained as well as the differences observed between both samples. Once more, we can confirm that higher conversion and therefore presence of a higher quantity of condensation products as well as heavier products are reached with TiO2 supported Au catalysts when compared with the ZnO catalysts of similar particle size. However, carbon deposits on ZnO catalysts consist of lighter physisorbed byproducts (diethyl ether, ethyl acetate, etc.), and the absence of condensed aromatic compounds avoid severe deactivation.
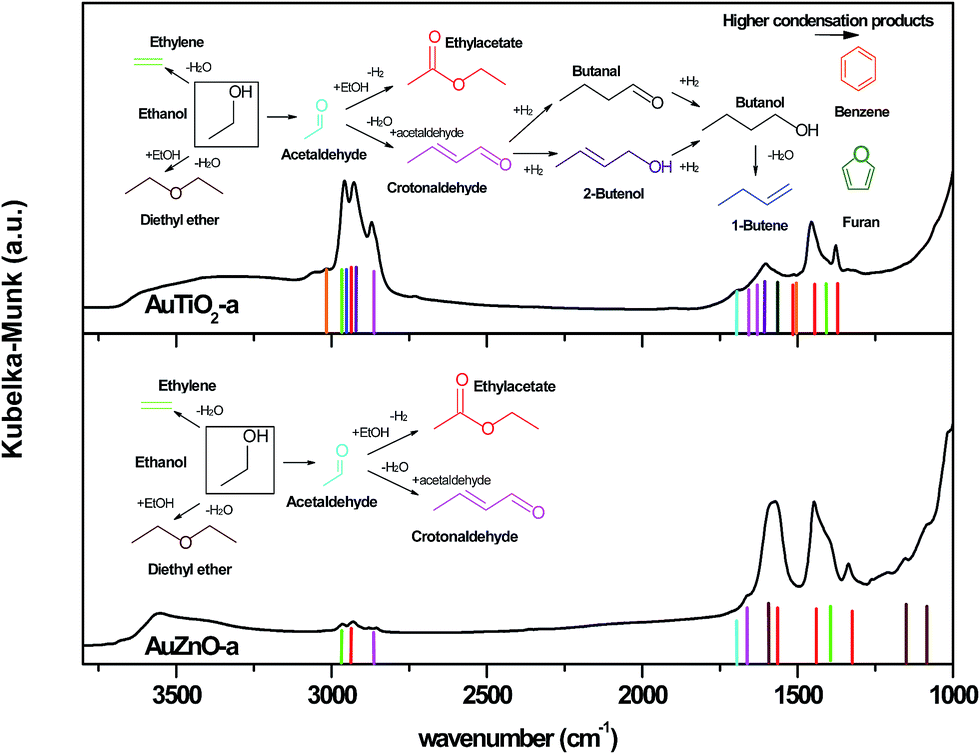 | ||
| Fig. 9 DRIFTS spectra of the two supported Au catalysts of similar particle size after reaction and identification of possible products adsorbed at the catalysts surface. | ||
Now, coming back to the fact that two catalysts of very similar particle size (AuTiO2-a: 2.2 and AuZnO-a: 2.6 nm) and metal loading (about 2% wt) would exhibit such different behavior in this catalytic test, as refers to conversion and product distribution, deserves additional discussion. As a matter of fact, it can only be attributed to their sole difference, the corresponding metal oxide supports. The support could exert influence in many ways without discarding any of its inherent properties such as specific surface area, redox or acid–base properties. Related to this latter are the pHPZC values which are reported to be 6.4 (ref. 58) and between 8.7–9.7 (ref. 59) at room temperature, for TiO2 and ZnO, respectively. The differences in the acid–base properties of the metal oxides, apart from causing effects on the deposition of the metal active phase would also participate in the selectivity towards the reaction products obtained as these supports also exhibit catalytic activity on their own. This is clearly visible on comparing the selectivity results depicted in Fig. 6 where while TiO2 was predominantly selective towards diethyl ether, acetaldehyde was the main product obtained with ZnO bare support. This has also been supported by literature where it is said that TiO2 offers higher selectivity towards diethyl ether in comparison to other oxides due to the fact that the stronger interaction between the reactant and surface leads to preferential formation of this product by the reaction between two molecules of ethanol.32 Likewise, in a previous study performed by our research group, acid and basic sites of ZnO materials were characterized by the isopropanol reaction test and confirmed that this material exposes mainly basic sites. This would explain the higher selectivity to the dehydrogenation product, acetaldehyde. At the same time, the formation of the byproduct, ethylene was conditioned by the presence of a specific type of acidic hydroxyl group located at the base surfaces of ZnO.45 Nevertheless, based on the significantly higher activity of noble metal compared with the bare support when employed as catalyst as we aforementioned, the support on its own would not cause these significant differences but rather would orient the differences observed in the resulting Au NPs deposition and therefore on the characteristics of this active phase exposed and responsible for the catalytic activity. In the previously cited ref. 32, the presence of Au changes the selectivity pattern of the TiO2 support due to its role promoting the ethanol dehydrogenation to acetaldehyde, and also favoring the hydrogen transfer step of aldol condensation product (view reaction scheme presented in Fig. 9) thereby resulting in the formation of C4 alcohols. This is in line with our obtained results. As for those of the ZnO series, the selectivity pattern was not changed with respect to that of the bare support but rather catalysts of very small nanoparticle sizes gave improved conversion values while the dehydrogenation product, acetaldehyde remained the most the principally obtained. Therefore, we cannot rule out the part played by nature of the support on the catalytic activity and selectivity of the active sites. This is clearly observed in our case where Au of similar particle sizes behaved so differently.
On the other hand, various studies involving Au NPs have pointed out the importance of strong metal-support interactions (SMSI).60–62 In a work published by Menegazzo et al. it was demonstrated how the properties of the support were decisive in the gold dispersion, position and shape.63 This was achieved by introducing sulphates on the ZrO2 surface. Their presence oriented the deposition of Au NPs in positions other than the corners and edges which were occupied by the sulphate groups. In another study of Akita et al., an atomic-scale structure investigation on the interface between Au(III) and γ-Fe2O3 was carried out by microscopy techniques and a preferential orientation relationship was observed between both materials.64 In our case, the differences in shapes and deposition sites could be induced by the characteristics of the two different metal oxides employed as support (TiO2 and ZnO). In a review article titled “Catalysis by gold: new insights into the support effect” emphasis is paid on the part played by the support in shaping the resulting supported Au NPs.65 Among other features discussed, the influence of reducibility of the metal oxide support is questioned. According to this review, TiO2 is considered under the category of reducible supports and tend to provide more favorable conditions for higher dispersion of hemispherically shaped NPs due to the stronger metal-support interaction. The consequent increase in interface between both materials is also said to be beneficial to catalytic activity. On the other hand, with irreducible oxides such as ZnO, lower interaction with the NPs is expected and thus, and although the presence of defects could cause modifications, spherically shaped particles are more likely to be formed. However, in a study carried out employing ZnO of different origins as support for Au NPs, it was said that in contrast to commercial nanocrystalline ZnO which allowed the deposition of more spherical particles, an epitaxial relation existed between the noble metal and the support when this latter was ZnO tetrapods.66 This enhanced its catalytic activity when compared to other ZnO conventional supports in the preferential oxidation reaction of carbon monoxide under hydrogen rich conditions. Nevertheless, and due to this epitaxial phenomenon, the interplanar distances of Au changed and lead to strain which was more accused for smaller particles causing the adverse effect of dractically reducing its catalytic performance.
From the above mentioned we can deduce that in our case, both supports would offer interaction with the Au particles but based on our results, it seems like the effects of this interactions would be more pronounced for those based on TiO2. This could be further confirmed taking into account characterization results previously discussed in the corresponding section where some Au1+ was detected (by XPS) in the TiO2 supported Au catalysts. This was not the case for the ZnO series. The oxidized gold species are said to diffuse at the interface between both materials.41 In addition, steps, corners and edges are known to be adsorbing sites in Au nanoparticles for certain molecules67 and the fraction of these sites would also be a consequence of the size and shape of Au NPs. All these could be applicable to our study where, the different shapes or positions obtained in both cases would explain why with one material higher activity and adsorption of certain reaction products is favored thereby leading to its deactivation. In other words, the selectivity and specially the catalytic activity of supported gold catalysts for the transformation reaction of ethanol could be controlled by tuning the size and morphology (degree of particle rounding and flattening, proportion of multiply twinned particles, number of steps, corners and edges, etc.) of the NPs as a consequence of the support chosen.
Conclusions
Our obtained results indicate that the catalytic properties of Au supported catalysts are strongly influenced by the support employed (type of metal oxide) and Au NP size (tuned during catalyst preparation). On one hand, within the same series of catalysts, Au nanoparticle size can be controlled by changing the synthesis temperature. When comparing both series of catalysts it is proved that TiO2 supported catalysts offered smaller particles and at the same time exhibited significantly higher initial conversion values, although these later decreased drastically especially in samples of smallest particle sizes. ZnO supported samples have more moderate initial conversions and in all cases deactivations were hardly observed even for samples with sizes as small as 2.6 nm. On the other hand, when comparing samples of similar particle size of each series, although in both cases the presence of gold facilitates the formation of acetaldehyde by dehydrogenation of ethanol, significant differences were clearly observed in terms of activity and product distribution. This fact proves the key role played by the support in the catalytic transformation of ethanol. The deactivation process has been evidenced (DRIFTS, XPS, TGA-MS) to be related to the presence of coke on the catalyst surface, inhibiting the activity. These carbon species consists of oxygenated hydrocarbons and aromatic compounds, the presence of the latter is more pronounced on the TiO2 supported samples, which explain the higher deactivation rate exhibited by these catalysts in comparison with the ZnO supported ones.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the Spanish Government (CTQ2014-52956-C3-2-R and -3-R, CTQ2017-89443-C3-1-R and -3-R projects). MVM is grateful to the UNED for a predoctoral grant.References
- M. Zhang and Y. Yu, Ind. Eng. Chem. Res., 2013, 52, 9505 CrossRef CAS.
- D. Fan, D.-J. Dai and H.-S. Wu, Materials, 2013, 6, 101 CrossRef CAS PubMed.
- E. V. Makshina, M. Dusselier, W. Janssens, J. Degrève, P. A. Jacobs and B. F. Sels, Chem. Soc. Rev., 2014, 43, 7917 RSC.
- J. H. Earley, R. A. Bourne, M. J. Watson and M. Poliakoff, Green Chem., 2015, 17, 3018 RSC.
- Y. Guan and E. J. M. Hensen, Appl. Catal., A, 2009, 361, 49 CrossRef CAS.
- A. Gazsi, A. Koos, T. Bansagi and F. Solymosi, Catal. Today, 2011, 160, 70 CrossRef CAS.
- V. Bolis, G. Cerrato, G. Magnacca and C. Morterra, Thermochim. Acta, 1998, 312, 63 CrossRef CAS.
- L. Ferretto and A. Glisenti, Chem. Mater., 2003, 15, 1181 CrossRef CAS.
- D. Stošić, S. Bennici, S. Sirotin, P. Stelmachowski, J.-L. Couturier, J.-L. Dubois, A. Travert and A. Auroux, Catal. Today, 2014, 226, 167 CrossRef.
- D. Stošić, S. Bennici, J.-L. Couturier, J.-L. Dubois and A. Auroux, Catal. Commun., 2012, 17, 23–28 CrossRef.
- N. Bou-Orm, A. Iorgu, S. Daniele and N. Guilhaume, Appl. Catal., A, 2013, 467, 414 CrossRef CAS.
- J. H. Kwak, J. Lee, J. Szanyi and C. H. F. Peden, Catal. Today, 2016, 265, 240 CrossRef CAS.
- T. Zaki, J. Colloid Interface Sci., 2005, 284, 606 CrossRef CAS PubMed.
- H. Zhou, J. Y. Wang, X. Chen, C.-L. O'Young and S. L. Suib, Microporous Mesoporous Mater., 1998, 21, 315 CrossRef CAS.
- J. Scalbert, F. Thibault-Starzyk, R. Jacquot, D. Morvan and F. Meunier, J. Catal., 2014, 311, 28 CrossRef CAS.
- N. Enjamuri, S. Hassan, A. Auroux, J. K. Pandey and B. Chowdhury, Appl. Catal., A, 2016, 523, 21 CrossRef CAS.
- J. I. Di Cosimo, V. K. Dıez, M. Xu, E. Iglesia and C. R. Apesteguıa, J. Catal., 1998, 178, 499 CrossRef CAS.
- H. Idriss, Platinum Met. Rev., 2004, 48, 105 CrossRef CAS.
- P. Osorio-Vargas, C. H. Campos, R. M. Navarro, J. L. G. Fierro and P. Reyes, Appl. Catal., A, 2015, 505, 159 CrossRef CAS.
- S. Y. Shen, T. S. Zhao and J. B. Xu, Int. J. Hydrogen Energy, 2010, 35, 12911 CrossRef CAS.
- C. Gonçalves Alonso, A. C. Furtado, M. Pereira Cantão, O. Aparecida Andreo dos Santos and N. Regina Camargo Fernandes-Machado, Int. J. Hydrogen Energy, 2009, 34, 3333 CrossRef.
- J. Fang, B. Zhang, Q. Yao, Y. Yang, J. Xie and N. Yan, Coord. Chem. Rev., 2016, 322, 1 CrossRef CAS.
- P. Johnston, N. Carthey and G. J. Hutchings, J. Am. Chem. Soc., 2015, 137, 14548 CrossRef CAS PubMed.
- A. M. Nadeem, G. I. N. Waterhouse and H. Idriss, Catal. Today, 2012, 182, 16 CrossRef CAS.
- W. Huang, in Heterogeneous Gold Catalysts and Catalysis, ed. Z. Ma and S. Dai, The Royal Society of Chemistry, Cambridge, UK, 2014, ch. 15, pp. 533–574 Search PubMed.
- T. Otto, S. I. Zones and E. Iglesia, J. Catal., 2016, 339, 195 CrossRef CAS.
- J. Fang, J. Li, B. Zhang, X. Yuan, H. Asakura, T. Tanaka, K. Teramura, J. Xie and N. Yan, Nanoscale, 2015, 7, 6325 RSC.
- Y. Kotolevich, E. Kolobova, E. Khramov, M. H. Farías, Ya. Zubavichus, H. Tiznado, S. Martınez-Gonzalez, V. Cortes Corberan, J. D. Mota-Morales, A. Pestryakov and N. Bogdanchikova, J. Mol. Catal. A: Chem., 2017, 427, 1 CrossRef CAS.
- E. del Río, D. Gaona, J. C. Hernández-Garrido, J. J. Calvino, M. G. Basallote, M. J. Fernández-Trujillo, J. A. Pérez-Omil and J. M. Gatica, J. Catal., 2014, 318, 119 CrossRef.
- V. I. Sobelev, O. A. Simakova and K. Y. Koltunov, ChemCatChem, 2011, 3, 1422 CrossRef.
- V. I. Sobelev, K. Y. Koltunov, O. A. Simakova, A.-R. Leino and D. Y. Murzin, Appl. Catal., A, 2012, 433–434, 88 CrossRef.
- J. Quesada, R. Arreola-Sanchez, L. Faba, E. Diaz, V. M. Renteria-Tapia and S. Ordoñez, Appl. Catal., A, 2018, 551, 23 CrossRef CAS.
- J. M. Hidalgoa, Z. Tislera, D. Kubickaa, K. Raabovab and R. Bulanek, J. Mol. Catal. A: Chem., 2016, 420, 178 CrossRef.
- J. F. DeWilde, C. J. Czopinski and A. Bhan, ACS Catal., 2014, 4, 4425 CrossRef CAS.
- R. Bacsa, J. Dexpert-Ghys, M. Verelst, A. Falqui, B. Machado, W. S. Bacsa, P. Chen, S. M. Zakeeruddin, M. Graetzel and P. Serp, Adv. Funct. Mater., 2009, 19, 875 CrossRef CAS.
- NIST Chemistry WebBook, NIST Standard Reference Database Number 69, http://webbook.nist.gov/chemistry/, accessed July 2016.
- Z. Ren, Y. Guo, Z. Zhang, C. Liua and P.-X. Gao, J. Mater. Chem. A, 2013, 1, 9897 CAS.
- M. Nasir Khan and J. Bashir, J. Mod. Phys., 2011, 2, 962 CrossRef.
- L. Guo, Y. L. Ji, H. Xu, P. Simon and Z. Wu, J. Am. Chem. Soc., 2002, 124, 14864 CrossRef CAS PubMed.
- E. Castillejos, E. Gallegos-Suarez, B. Bachiller-Baeza, R. Bacsa, P. Serp, A. Guerrero-Ruiz and I. Rodríguez-Ramos, Catal. Commun., 2012, 22, 79 CrossRef CAS.
- M. Ousmane, L. F. Liotta, G. Pantaleo, A. M. Venezia, G. Di Carlo, M. Aouine, L. Retailleau and A. Giroir-Fendler, Catal. Today, 2011, 176, 7 CrossRef CAS.
- N. Zheng and G. D. Stucky, J. Am. Chem. Soc., 2006, 128, 14278 CrossRef CAS PubMed.
- M. C. Holz, K. Tölle and M. Muhler, Catal. Sci. Technol., 2014, 4, 3495 CAS.
- D. Gabriëls, W. Y. Hernández, B. Sels, P. Van Der Voort and A. Verberckmoes, Catal. Sci. Technol., 2015, 5, 3876 Search PubMed.
- M. V. Morales, E. Asedegbega-Nieto, A. Iglesias-Juez, I. Rodriguez-Ramos and A. Guerrero-Ruiz, ChemSusChem, 2015, 8, 2223 CrossRef CAS PubMed.
- E. Santacesaria, G. Carotenuto, R. Tesser and M. Di Serio, Chem. Eng. J., 2012, 179, 209 CrossRef CAS.
- P. Castaño, G. Elordi, M. Olazar, A. T. Aguayo, B. Pawelec and J. Bilbao, Appl. Catal., B, 2011, 104, 91 CrossRef.
- Y. Li, C. Zhang, Y. Liu, S. Tang, G. Chen, R. Zhang and X. Tang, Fuel, 2017, 189, 23 CrossRef CAS.
- R. Fiorenza, M. Bellardita, L. Palmisano and S. Scirè, J. Mol. Catal. A: Chem., 2016, 415, 56 CrossRef CAS.
- D. Lin-Vien, N. Colthup, W. Fateley and J. Grasselli, The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules, Academic Press, 1991, ISBN: 978-0-12-451160-6 Search PubMed.
- H. De Paz-Simon, A. Chemtob, C. Croutxé-Barghorn, S. Rigolet, L. Michelin, L. Vidal and B. Lebeau, Langmuir, 2013, 29, 1963 CrossRef CAS PubMed.
- R. Kydd, W. Y. Teoh, J. Scott, D. Ferri and R. Amal, ChemCatChem, 2009, 1, 286 CrossRef CAS.
- A. Yee, S. J. Morrison and H. Idriss, J. Catal., 2000, 191, 30 CrossRef CAS.
- C. Drouilly, J.-M. Krafft, F. Averseng, H. Lauron-Pernot, D. Bazer-Bachi, C. Chizallet, V. Lecocq and G. Costentin, Catal. Today, 2013, 205, 67 CrossRef CAS.
- R. Yang, Y. Fu, Y. Zhang and N. Tsubaki, J. Catal., 2004, 228, 23 CrossRef CAS.
- R. Yang, Y. Zhang and N. Tsubaki, Catal. Commun., 2007, 8, 1829 CrossRef CAS.
- J. Llorca, N. Homs and P. Ramirez de la Piscina, J. Catal., 2004, 227, 556 CrossRef CAS.
- N. Kallay, T. Madić, K. Kučej and T. Preočanin, Colloids Surf., A, 2003, 230, 3 CrossRef CAS.
- C. B. Tay, S. J. Chua and K. P. Loh, J. Cryst. Growth, 2009, 311, 1278 CrossRef CAS.
- A. A. Herzing, C. J. Kiely, A. F. Carley, P. Landon and G. J. Hutchings, Science, 2008, 321, 1331 CrossRef CAS PubMed.
- Z. Wang, H. Fu, Z. Tian, D. Hana and F. Gu, Nanoscale, 2016, 8, 5865 RSC.
- P. H. Rana and P. A. Parikh, J. Ind. Eng. Chem., 2017, 47, 228 CrossRef CAS.
- F. Menegazzo, M. Signoretto, D. Marchese, F. Pinna and M. Manzoli, J. Catal., 2015, 326, 1 CrossRef CAS.
- T. Akita, Y. Maeda and M. Kohyama, J. Catal., 2015, 324, 127 CrossRef CAS.
- X. Y. Liu, A. Wang, T. Zhang and C.-Y. Mou, Nano Today, 2013, 8, 403–416 CrossRef CAS.
- E. Castillejos, R. Bacsa, A. Guerrero-Ruiz, I. Rodriguez-Ramos, L. Datas and P. Serp, Nanoscale, 2011, 3, 929 RSC.
- M. Haruta and M. Daté, Appl. Catal., A, 2001, 222, 427 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra00314a |
| This journal is © The Royal Society of Chemistry 2018 |