
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanofiller-conjugated percolating conductive network modified polymerization reaction characteristics of aromatic thermosetting copolyester resin†
Mete Bakir a,
Jacob L. Meyerad,
Andre Sutrisnoc,
James Economybd and
Iwona Jasiuk*a
a,
Jacob L. Meyerad,
Andre Sutrisnoc,
James Economybd and
Iwona Jasiuk*a
aDepartment of Mechanical Science and Engineering, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA. E-mail: ijasiuk@illinois.edu; Tel: +1-217-333-92-59
bDepartment of Materials Science and Engineering, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA
cNMR/EPR Laboratory, School of Chemical Sciences, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA
dATSP Innovations, Champaign, IL 61820, USA
First published on 29th January 2018
Abstract
Deliberately controlled interfacial interactions between incorporated nanofiller particles and host polymer backbone chains constitute a critical element in the realm of polymer nanocomposites with tailorable multifunctional properties. We demonstrate the physicochemical effects induced by graphene nanoplatelets (GNP) of different sizes on the condensation polymerization reaction of aromatic thermosetting copolyester (ATSP) through the formation of electrically conductive percolating networks as enabled by interfacial interactions. Carboxylic acid and acetoxy-capped precursor oligomers of ATSP are solid-state mixed with chemically pristine GNP particles at various loading levels. Upon in situ endothermic condensation polymerization reaction, crosslinked backbone of the ATSP foam matrix is formed while the carbonaceous nanofillers are incorporated into the polymer network via covalent conjugation with functional end-groups of the oligomers. The controlled GNP size promotes different electrical percolation thresholds and ultimate electrical conductivities. Microstructural analysis demonstrates GNP distributions in the matrix as well as morphological modifications induced by the formation of conductive percolating GNP networks. Cure characteristics reveal the thermochemical changes prompted in the polymerization processes for GNP content above the requirement for percolation formation. Chemical spectroscopy of the ATSP nanocomposite morphology exhibits the formation of a robust interfacial coupling mechanism between the GNPs and ATSP backbone. The findings here may guide the developmental efforts of nanocomposites through better identifying roles of the morphology and content of nanofillers in polymerization processes.
1. Introduction
Technological advancements of structural materials currently count on the development of alternative lightweight materials possessing physical properties commensurate with contemporary systems while enabling cost-effective and industrially scalable high-throughput production solutions.1–4 Polymer nanocomposites are combinations of nanofiller reinforcement particles and host polymer matrix, which uniquely embody multifunctional properties within low-density morphologies.5–7 Physical properties of the polymer nanocomposites can be adjusted by controlling the nanofiller loading at large. Yet, optimization of such properties involves many factors coming into effect including but not limited to processing conditions of the nanocomposites as well as morphology, dispersion quality, and chemical functionalization of the nanofillers.8–11 In this regard, recent efforts have sought to realize robust interfacial attachment schemes between edge-or-surface-functionalized nanofillers and backbone chains of various polymers.12 Yet, current nanocomposite configurations are limited in terms of their ultimate physical properties due to the presence of only weak intermolecular coupling mechanisms.13In particular, electrically conductive polymer nanocomposites have been a subject of extensive studies owing to their immense potential for broad spectrum of applications spanning batteries, membranes, and electromagnetic shielding.14–16 Within this framework, the formation of conductive networks of interconnected nanofillers – past the electrical percolation transition threshold – within intrinsically insulating polymer domains is imperative as far as the performances of the electrically conductive nanocomposites are concerned.17 The percolation threshold is recognized to vary with respect to morphology, size, and distribution of nanofillers as well as rheological characteristics of resins.18 On the other hand, it is well acknowledged that the increased nanofiller loading fractions, near or above the percolation transition, substantially deteriorate the structural integrity of the nanocomposites.19 Hence, a primary research thrust has been centered on efforts to realize minimal loading levels for percolation thresholds utilizing various types of nanofillers and polymer combinations prepared via different processing methods.20,21 Although, there are comprehensive studies reporting on the rheological properties of the nanocomposites as a result of incremented nanofiller contents, little is still known about the physicochemical effects of the nanofillers on characteristics of in situ polymerization processes.22
Aromatic thermosetting copolyester (ATSP) utilizes low cost, easily processable and crosslinkable oligomers to develop a high-performance polymer morphology (Scheme 1).23 The tailorable chemical structure of the backbone chain as well as convenient reconfigurability of the matrix into various form factors enable effectively controllable physical properties.24 Recently, ATSP nanocomposites have been introduced that facilitate improved distributions of nanofillers via a solid-state mixing route, which then result in substantially increased thermal and mechanical properties.5 In this work, we report on the physicochemical changes observed during in situ polycondensation reaction through formation of electrically conductive percolating GNP networks within the ATSP resin. ATSP nanocomposite foams are obtained through a polycondensation reaction between carboxylic acid- and acetoxy-capped constituent oligomers, which releases acetic acid as a reaction by-product to generate porous morphology. In the course of the in situ polymerization reaction, the GNPs covalently conjugate with the functionalized oligomers while the resin is advancing in molecular weight to develop the foam structures. The varied GNP size controls the electrical percolation transition thresholds and measured ultimate electrical conductivities. As well, microstructural analysis displays the morphological effects caused by the percolating networks of the GNPs. Cure characteristics reveal the influence of the nanofillers on the polymerization reaction. Chemical characterization of the nanocomposite backbone displays strong interfacial entanglement of the GNPs and ATSP matrix.
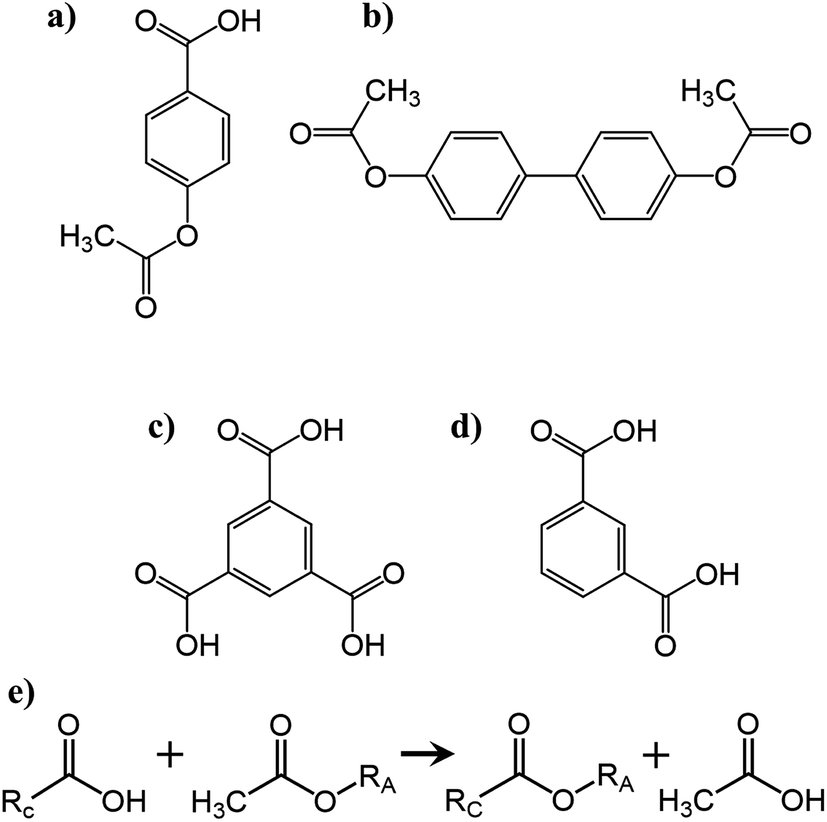 | ||
| Scheme 1 Chemical structures of the monomers (a) 4-acetoxybenzoic acid (ABA), (b) biphenol diacetate (BDPA), (c) trimesic acid (TMA) and (d) isophthalic acid (IPA). (e) Chemical representation of the polycondensation reaction carried out between the acetoxy functionalized oligomer (RA) (Fig. S1†) and carboxylic acid functionalized oligomer (RC) (Fig. S2†) yielding a crosslinked polymer backbone and releasing acetic acid as a reaction by-product. | ||
2. Materials and experimental methods
2.1. Fabrication of aromatic thermosetting copolyester nanocomposites
The constituent carboxylic acid and acetoxy functional group matching oligomers of the aromatic thermosetting copolyester are synthesized using biphenol diacetate (BPDA), 4-acetoxybenzoic acid (ABA), isophthalic acid (IPA), and trimesic acid (TMA) monomers (Sigma-Aldrich Co., USA) at particular molar feed ratios as further explained in earlier studies.23–25 The carboxylic acid (∼1201 g mol−1) and acetoxy-capped (∼1203 g mol−1) oligomers (Fig. S1, S2 and Table S1†) (premixed at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 weight ratio) are combined in solid state with chemically pristine (non-functionalized) graphene nanoplatelet (GNP) particles of 1 μm (thickness: 8–15 nm) (Cheap Tubes, Inc., USA), 5 μm (thickness: 6–8 nm) (XG Sciences, Inc., USA), and 25 μm (thickness: 6–8 nm) (XG Sciences, Inc., USA) average flake diameters at 1, 3, 4, 5, 6, 7, 8, and 10 wt% loading levels.5 The GNPs and ATSP precursor oligomers mixtures form powder combinations in solid state. The mixing process is performed via rigorous shaking until white oligomers powder turns to pitch dark, as also investigated under UV light. Such a color change indicates effective distribution of the nanofillers at macroscale in the oligomer powder bed.25 The ATSP nanocomposites are obtained via condensation polymerization (polycondensation) reaction between the constituent oligomers, which generates a crosslinked aromatic polyester backbone polymer network.25 The thermal polymerization process includes two dwell stages at 202 °C for 90 minutes and 270 °C for 150 minutes, respectively. The cycle has a final cure stage at 330 °C for 90 minutes.5 The nanocomposite foams are labeled as ATSP-GNP type as their naming convention.
:1 weight ratio) are combined in solid state with chemically pristine (non-functionalized) graphene nanoplatelet (GNP) particles of 1 μm (thickness: 8–15 nm) (Cheap Tubes, Inc., USA), 5 μm (thickness: 6–8 nm) (XG Sciences, Inc., USA), and 25 μm (thickness: 6–8 nm) (XG Sciences, Inc., USA) average flake diameters at 1, 3, 4, 5, 6, 7, 8, and 10 wt% loading levels.5 The GNPs and ATSP precursor oligomers mixtures form powder combinations in solid state. The mixing process is performed via rigorous shaking until white oligomers powder turns to pitch dark, as also investigated under UV light. Such a color change indicates effective distribution of the nanofillers at macroscale in the oligomer powder bed.25 The ATSP nanocomposites are obtained via condensation polymerization (polycondensation) reaction between the constituent oligomers, which generates a crosslinked aromatic polyester backbone polymer network.25 The thermal polymerization process includes two dwell stages at 202 °C for 90 minutes and 270 °C for 150 minutes, respectively. The cycle has a final cure stage at 330 °C for 90 minutes.5 The nanocomposite foams are labeled as ATSP-GNP type as their naming convention.
2.2. Physical characterization of aromatic thermosetting copolyester nanocomposites
Solid-state nuclear magnetic resonance spectroscopy measurements are performed using ground nanocomposite specimens (∼50 mg) packed into NMR rotors with Unity Inova 300 MHz NMR spectrometer (Varian Inc., USA). 1H and 13C spectra are acquired using direct pulse (DP) (pulse width (pw) = 2.5 μs, recycle time (d1) = 2 s) and cross-polarized (CP) (pulse width (pwH) = 2 μs, recycle time (d1) = 2 s, and contact time (tHX) = 4 ms) excitations, respectively. Specimens are spun at 10 kHz. Data analysis is performed using the MestreNova software. 1H and 13C NMR spectra are processed using 1 Hz and 25 Hz line broadenings, respectively.Scanning Electron Microscopy (SEM) (S-4800, Hitachi, Japan) is operated in the high-resolution mode (10 kV voltage and 5 μA current) to image microstructural features and distributions of the GNPs in the ATSP matrix.
Transmission electron microscopy is employed in bright-field mode (200 kV, 102 μA) to analyze the interfacial interactions of the GNPs with the ATSP chains (2010LaB6, JEOL, Japan). Fabricated nanocomposite specimens are ground using a laboratory grinder and the powder is settled in methanol solution for 10–15 min. Floating particles in the solution are collected using a pipette and passed through a copper grid, which is then dried at 80 °C for 1 hour.
Raman spectroscopy (Raman 11, Nanophoton, Japan) measurements are carried out using an excitation source of 633 nm, a 20× objective lens, and a total acquisition time of 5 min.
Direct current (DC) electrical conductivity measurements are performed on foam morphology specimens using 4 point-probe method (6517 B, Keithley Instruments, USA). The specimens are in 5 × 5 × 12 mm3 (width × thickness × length). The DC electrical conductivity results are averaged over four samples per loading fraction, and standard deviations are given by error bars, accordingly. For the loading levels corresponding to percolation transitions, the nanocomposite specimens are labeled, in addition to the GNP size, with <φc, ∼φc, >φc denoting below percolation, around percolation, and above percolation GNP contents, respectively.
Volumetric microstructural images are obtained on foam morphology specimens using a high-resolution X-ray micro-computed tomography (Micro-CT) (Xradia MicroXCT-400). The 3D objects are reconstructed utilizing total of 1441 images taken at every 0.25° corresponding to sample rotation during the imaging process. The samples are in 10 mm × 20 mm cross-section with 4 mm thickness.
Density is calculated as the ratio of measured weight to volume of the specimens. The density of neat fully dense ATSP is 1.27 Mg m−3.
The cure characteristics of the nanocomposite foams are investigated using a Differential Scanning Calorimetry (DSC) (DSC 2910, TA Instruments, USA). The tests are performed under an inert atmosphere of nitrogen. The cure cycle involves a temperature-ramped heating process with a 10 °C min−1 heating rate.
3. Results and discussion
3.1. Polymerization reaction and interfacial coupling mechanism
First, we highlight the chemical principles of the polycondensation reaction carried out between the matching oligomer groups that form the crosslinked network of the ATSP matrix. The polymerization process involves the acetoxy and carboxylic acid-capped constituent oligomers conducting an esterification condensation reaction at sufficiently high temperatures exceeding 200 °C (for chemical structures of the oligomers see Fig. S1 and S2†). In the course of this reaction, ether oxygen groups of the acetoxy-capped oligomer exchange with the hydroxyl groups of the carboxylic acid-capped oligomer that produces the crosslinked aromatic backbone morphology while releasing acetic acid as a by-product, which consequently generates a porous morphology.25 Fig. 1 demonstrates solid-state Nuclear Magnetic Resonance (ssNMR) spectra of the individual carboxylic acid (C-group)- and acetoxy (A-group)-capped oligomer groups, uncured combination of the oligomers (C + A mixed), and cured ATSP matrix (ATSP). In Fig. 1a, 13C cross-polarization magic-angle spinning (CPMAS) spectra of the individual oligomers exhibit two main peak domains corresponding to aromatic main chains (C–C/C–H bonds) and functional side chains (C–O/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds). Measured chemical shift positions of the C-group are δ = 171.7, 162.9, 154.8, 150.5, 134.9, 129.9, and 122.1 ppm and A-group are δ = 168.7, 162.8, 154.8, 150.5, 136.2, 131.2, 127.3, 122.1, and 20.6 ppm. The solid-state uncured combination of the oligomers (C + A mixed) displays similar characteristic peaks detected at δ = 172.9, 168.9, 162.9, 154.8, 150.5, 135.2, 129.9, 122.1, and 20.6 ppm. Following the polymerization reaction (performed outside of the NMR station), the cured ATSP morphology exhibits peaks at δ = 160.5, 152.6, 148.2, 132.9, 128.4, 125.2, 118.8 ppm. Hence, the two identified peak domains, emerging from the constituent oligomer groups, are effectively preserved within the ATSP morphology whereby a crosslinked aromatic backbone linked via oxygen bonds is formed.25 Regarding the chemical imprints of the reactive caps, the peaks observed at δ = 171.7 ppm in C-group, and δ = 168.7 and 20.6 ppm in A-group (as indicated with asterisks in the figure) likewise transmit to the spectrum of the uncured mixture, yet disappear upon polymerization, and are absent from the 13C spectrum of the cured ATSP matrix. It highlights that these peaks are associated with the reactive caps that participate in the reaction. In particular, the ether oxygen of the acetoxy cap (with the methyl (–CH3) group displays at δ = 20.6 ppm and the oxygen-bearing groups (C–O/CO) display at δ = 168.7 ppm as observables) interacts with the hydroxyl group of the carboxylic acid cap (with the oxygen-bearing groups (C–O/CO) show a peak at δ = 171.7 ppm) in the course of the polycondensation reaction which then releases acetic acid foaming as the by-product while the crosslinked morphology is formed.26 Besides, in Fig. 1b, 1H direct pulse magic-angle spinning (DPMAS) spectra show chemical shifts of the C-group at δ = 7.4 ppm, A-group at δ = 7.4, 1.7 ppm, C + A mixed at δ = 7.4, 2.1 ppm, and ATSP at δ = 5.0 ppm (the shifts are referenced to zero in the figure for clarity). Particularly, the spectrum of the A-group displays a shoulder formation (at δ = 1.7 ppm) over the main peak, which represents the acetoxy based reactive cap of the oligomer.27 Note that although the acetoxy cap is detected in the 1H spectrum of the uncured mixture, it is no longer observable after the esterification process. Hence, based on both 1H and 13C spectra, the formation of the crosslinked aromatic backbone is mainly controlled by the functional groups of the constituent oligomers reacting at elevated temperatures during the polymerization reaction.
O bonds). Measured chemical shift positions of the C-group are δ = 171.7, 162.9, 154.8, 150.5, 134.9, 129.9, and 122.1 ppm and A-group are δ = 168.7, 162.8, 154.8, 150.5, 136.2, 131.2, 127.3, 122.1, and 20.6 ppm. The solid-state uncured combination of the oligomers (C + A mixed) displays similar characteristic peaks detected at δ = 172.9, 168.9, 162.9, 154.8, 150.5, 135.2, 129.9, 122.1, and 20.6 ppm. Following the polymerization reaction (performed outside of the NMR station), the cured ATSP morphology exhibits peaks at δ = 160.5, 152.6, 148.2, 132.9, 128.4, 125.2, 118.8 ppm. Hence, the two identified peak domains, emerging from the constituent oligomer groups, are effectively preserved within the ATSP morphology whereby a crosslinked aromatic backbone linked via oxygen bonds is formed.25 Regarding the chemical imprints of the reactive caps, the peaks observed at δ = 171.7 ppm in C-group, and δ = 168.7 and 20.6 ppm in A-group (as indicated with asterisks in the figure) likewise transmit to the spectrum of the uncured mixture, yet disappear upon polymerization, and are absent from the 13C spectrum of the cured ATSP matrix. It highlights that these peaks are associated with the reactive caps that participate in the reaction. In particular, the ether oxygen of the acetoxy cap (with the methyl (–CH3) group displays at δ = 20.6 ppm and the oxygen-bearing groups (C–O/CO) display at δ = 168.7 ppm as observables) interacts with the hydroxyl group of the carboxylic acid cap (with the oxygen-bearing groups (C–O/CO) show a peak at δ = 171.7 ppm) in the course of the polycondensation reaction which then releases acetic acid foaming as the by-product while the crosslinked morphology is formed.26 Besides, in Fig. 1b, 1H direct pulse magic-angle spinning (DPMAS) spectra show chemical shifts of the C-group at δ = 7.4 ppm, A-group at δ = 7.4, 1.7 ppm, C + A mixed at δ = 7.4, 2.1 ppm, and ATSP at δ = 5.0 ppm (the shifts are referenced to zero in the figure for clarity). Particularly, the spectrum of the A-group displays a shoulder formation (at δ = 1.7 ppm) over the main peak, which represents the acetoxy based reactive cap of the oligomer.27 Note that although the acetoxy cap is detected in the 1H spectrum of the uncured mixture, it is no longer observable after the esterification process. Hence, based on both 1H and 13C spectra, the formation of the crosslinked aromatic backbone is mainly controlled by the functional groups of the constituent oligomers reacting at elevated temperatures during the polymerization reaction.
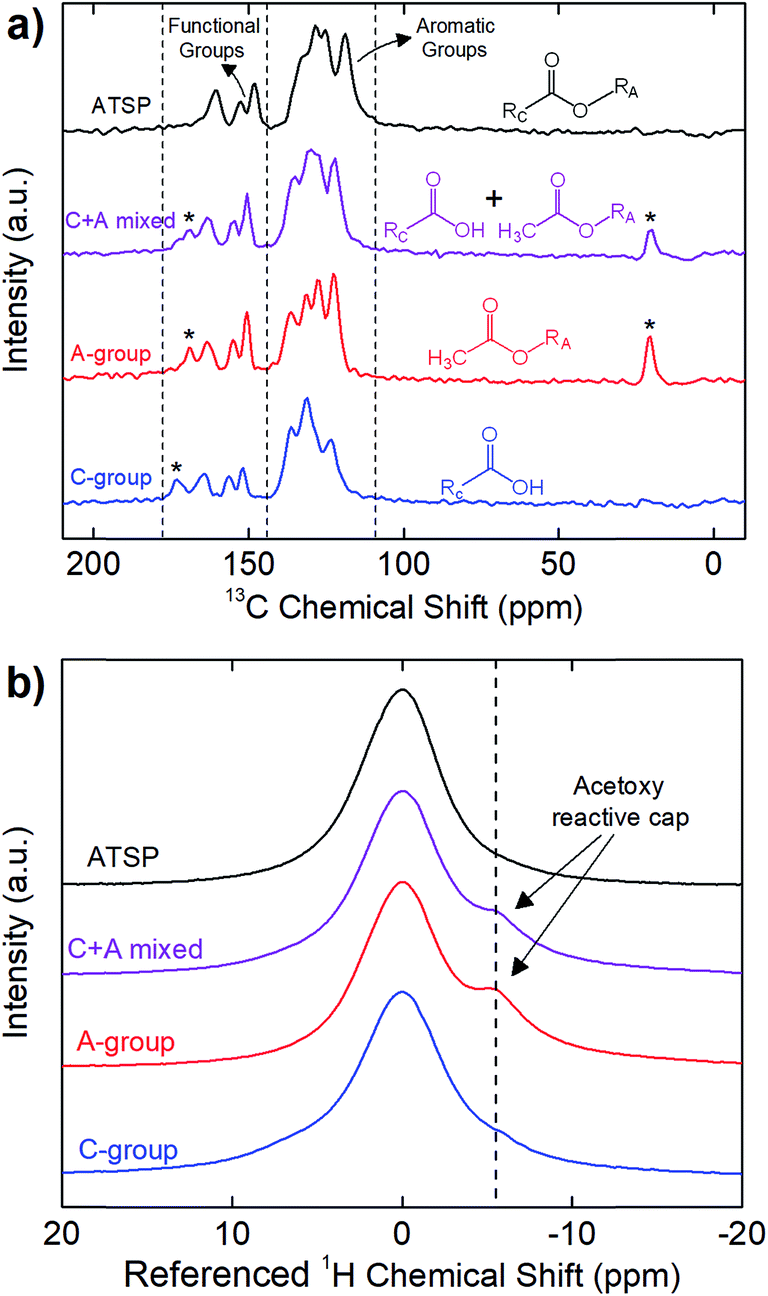 | ||
| Fig. 1 Solid-state nuclear magnetic resonance (ssNMR) 13C cross-polarization magic-angle spinning (CPMAS) (a) and 1H direct pulse magic-angle spinning (DPMAS) (b) spectra of the carboxylic acid (C-group) and acetoxy (A-group) capped individual oligomers, uncured combination of the C- and A-groups (C + A mixed), and cured matrix (ATSP). Aromatic groups and functional groups represent C–C/C–H and C–O/CO bonds participated in the oligomer sets as well as the cured matrix. RC and RA indicate aromatic chain configurations of the carboxylic acid and acetoxy group oligomers, respectively. Asterisks highlight the peaks associated with the reactive caps involving in the polymerization reaction. 1H spectra referenced to zero to display the additional peak formations, corresponding chemical shift values are given in the text. | ||
Next, we demonstrate the in situ intermolecular attraction and interfacial coupling mechanism effective between the GNP nanoparticles and ATSP backbone chains, as shown in Fig. 2. During the thermal polymerization reaction between the constituent oligomers, the GNP particles interact with the reactive functional groups of the oligomers, and consecutively integrate with the crosslinked network upon curing. The underlying in situ interaction mechanism stems from intermolecular attraction forces between highly polar acetoxy and carboxylic acid caps of the oligomers and inherently oxygen-containing polar sites on the GNP particles.28 Within this scheme, molten oligomers at high temperatures blend together and wet the GNP particles through effective hydrodynamic forces emerging during the acetic acid by-product release.5 It then facilitates the polar force induced interfacial interaction between the GNPs and ATSP. Subsequently, the oligomers – containing acetoxy and carboxylic acid reactive groups – crosslink with the oxygen-bearing polar sites on the GNP particles upon the cure process which effectively tethers the GNPs to the crosslinked domain of the ATSP. Since the interfacial coupling occurs through oxygen bonds, it enables high physicochemical stability which effectively enhances the thermomechanical properties of the ATSP nanocomposites.25 Scanning electron microscopy (SEM) analysis reveals that the GNP particles are thickly coated by the matrix indicating effective surface wettability enabled through the in situ attraction mechanism (Fig. 2a). Hence, the GNP particles are not phobic to the molten oligomer groups during the polymerization process. More importantly, such an intrinsic interaction scheme indicates that the GNPs can also modify the polymerization reactions characteristics, which will be discussed later in the text. To highlight the interfacial coupling effect, transmission electron microscopy (TEM) image displays a GNP flake having ATSP chain fragments (darker domains) tethered on the surface that clearly indicates the molecular level extent of the coupling formation (Fig. 2b). In addition, Raman spectroscopy measurements, as shown in Fig. 2c, display the pristine GNP spectrum as compared to that of the GNP interacted with the ATSP matrix. The pristine GNP produces a conventional spectrum with two characteristic peaks at 1360 cm−1 and 1590 cm−1 which correspond to D (disordered hybridized structure with impurities) and G (ordered graphitic structure) bands, respectively.29 Upon the GNP particles interacting with the ATSP backbone chains, additional peak formations are observed, as denoted with asterisks. The peaks arise from the polymer domain for which the polymer chains are excited while strongly attached to the GNP structure (see Fig. S3† for Raman spectrum of the neat ATSP). Also note that the presence of the additional peaks disturbs the D band region, which indicates that the grafting takes place through a disordered area of the GNP flakes where the oxygen-containing sites are present. Hence, it is clearly evident that the GNP and ATSP matrix constitute a strong interfacial attachment. We note that the GNP particles remained intact in the ATSP matrix upon the polymerization reaction which apparently did not exfoliate, based on X-ray diffraction (XRD) spectroscopy demonstrated in an earlier work.5 Additionally, temperature-driven hydrodynamic motion within molten oligomer domain caused relocation of the acetic acid blowing agent bubbles enabling redistribution and rearrangement of the GNP particles prior to the curing that minimized nanoparticle aggregation in the nanocomposite matrix.5
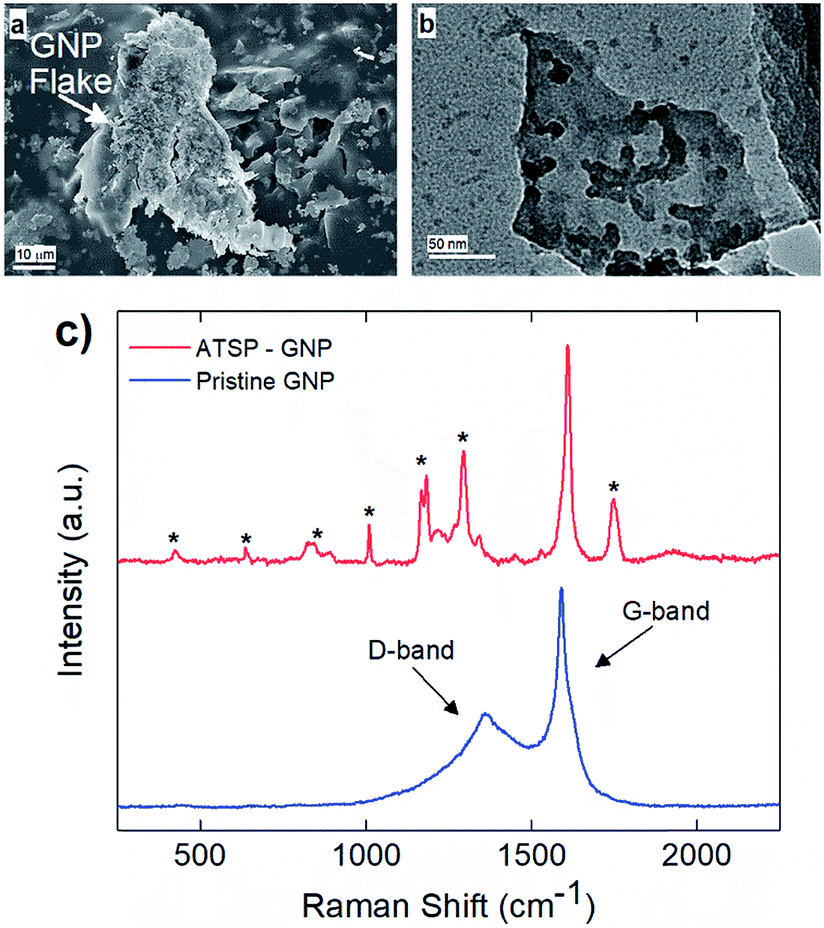 | ||
| Fig. 2 A scanning electron microscopy image obtained on a single GNP flake (GNP 25 μm) thickly coated by ATSP matrix (a). A bright-field transmission electron microscopy image of a GNP flake (GNP 5 μm) decorated with interfacially tethered ATSP chain fragments (b). Raman spectra of pristine GNP flakes (GNP 5 μm) and GNPs within the ATSP matrix (c). Asterisks denote additional peaks formed upon interactions with the ATSP backbone chains. | ||
3.2. Electrical percolation characteristics
Afterwards, we investigate the electrical percolation characteristics of the ATSP nanocomposites incorporated with the GNP particles of 1 μm, 5 μm, and 25 μm sizes. Direct Current (DC) electrical conductivity results of the ATSP nanocomposites obtained employing a four-point probe method are shown in Fig. 3. The neat ATSP (without addition of the GNPs) displays a characteristic non-conductive behavior with a DC conductivity of 4.04 × 10−13 ± 7.73 × 10−14 S m−1 being in agreement with similar polymer systems.30 Upon selectively incremented nanofiller content, the electrical conductivity remains almost constant until the percolation threshold transitions of each GNP sizes. The percolation thresholds are observed to vary with respect to the GNP particle sizes. In particular, GNP 1 μm forms the conductive percolating network around 5 wt% while GNP 5 μm and GNP 25 μm obtain the interconnected domains near 4 wt% and 3 wt%, respectively. The percolation thresholds are visually determined with respect to the S-shape characteristic curve of the percolation transition. The observed percolation thresholds concur with the findings in the literature.31–33 The downshifts in the percolation thresholds with the larger GNPs occur due to the prompt development of the percolating networks which increases the probability of the formation of nanoparticle conductive paths in the matrix.34 The generalized connectedness percolation theory describes that the percolation threshold is independent of the aspect ratio for monodisperse disc-shaped platelet nanofillers.17,35 Yet, from an experimental point of view, difference in the average diameters of the GNPs (polydispersity) is expected for each nanocomposite, although they are formed through a single GNP size, considering the bulk manufacturing processes of the pristine GNPs as well as the processing conditions of the nanocomposites. Hence, the shifts in the percolation thresholds with respect to different GNP sizes are conceivable. In this regard, as polydispersity ratio (the variation of the GNP size for a certain given diameter) is increased, in this case through the presence of the larger GNP particles (assuming same thicknesses for all sizes), the percolation threshold decreases.17 Above the percolation transitions, the nanocomposites acquire highly conductive states. As a matter of fact, we observe that the 10 wt% of GNP loading level displays electrical conductivities of 4.08 × 10−3 ± 1.81 × 10−3 S m−1 for GNP 1 μm, 21.2 ± 9.08 S m−1 for GNP 5 μm, and 1.6 × 103 ± 4.78 102 S m−1 for GNP 25 μm. A possible reason which gives rise to lower electrical conductivities with the smaller aspect ratio of the GNPs is the increased interfacial contact resistance through larger contact area.36 Another reason may be the different intrinsic electrical conductivities for each different GNP size induced through the presence of inherent functional groups or defect sites, yet conclusive remarks require further analysis. Based on the electrical conductivity results, we determine 3 percolation-based GNP contents for further physical analysis: below percolation (<φc), around percolation (∼φc), and above percolation (>φc). For GNP 1 μm, <φc, ∼φc, >φc correspond to 1 wt%, 5 wt%, and 10 wt%, respectively. For GNP 5 μm, <φc, ∼φc, >φc correspond to 1 wt%, 4 wt% and 10 wt%, respectively. For GNP 25 μm, <φc, ∼φc, >φc correspond to 1 wt%, 3 wt%, and 10 wt%, respectively.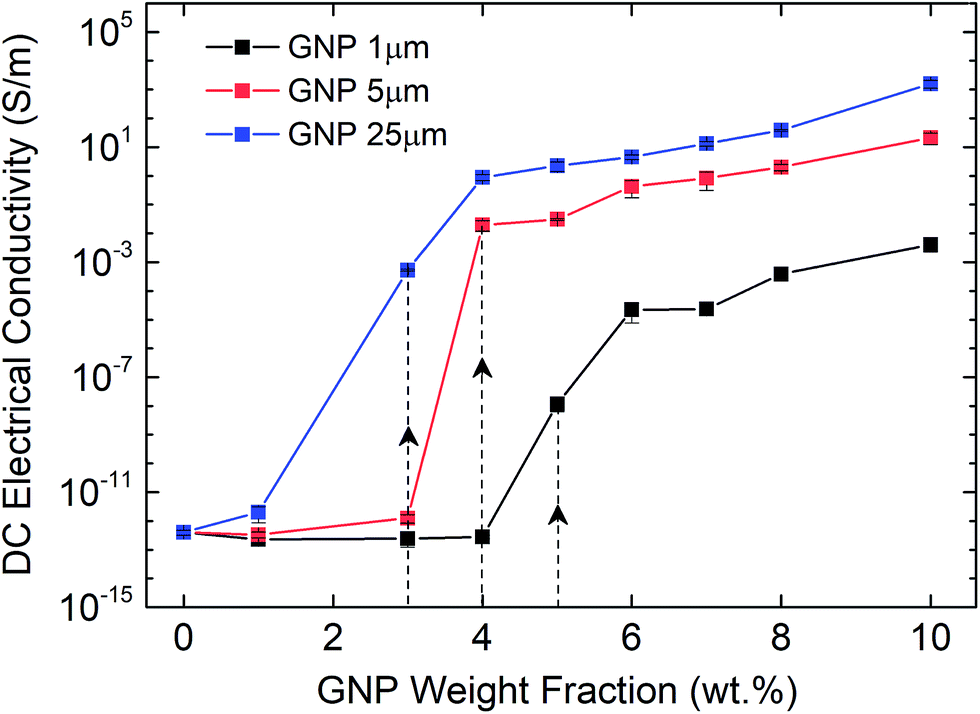 | ||
| Fig. 3 Results of DC electrical conductivity measurements on the ATSP-GNP nanocomposites with GNPs of 1 μm, 5 μm, and 25 μm. Standard deviations were represented with error bars. Dashed lines indicate assigned percolation thresholds for each GNP size. | ||
To further interrogate the percolation characteristics, we perform microstructural analysis via a Scanning Electron Microscope (SEM) on a set of specimens with GNP contents of below percolation (<φc), around percolation (∼φc), and above percolation (>φc). As shown in Fig. 4a–c, GNP 1 μm exhibits very random and wide distributions until the percolation threshold after which the GNP flakes are observed to effectively cover the surface of the matrix. In addition, in Fig. 4d–f, GNP 5 μm specimens are observed to form some small clusters below the percolation point owing to the larger particle size. Upon increases in the content, the GNPs are embedded in the matrix. As well, above the percolation transition, the GNP particles are thickly coated with ATSP while dispersed over the matrix. In Fig. 4g–i, GNP 25 μm specimens display similar characteristics as GNP 5 μm, yet due to the larger particle size formations of cluster islands are clearly marked. The findings here indicate that the GNPs of the larger sizes (5 μm and 25 μm) form cluster through percolation formation, significantly interact with the matrix, and effectively alter the topological features of the nanocomposite morphologies. Also, from a modeling perspective, the establishment of the percolating networks through the formation of clusters (defined as connectedness) for the larger GNPs significantly affects the percolation transition characteristics as well as the ultimate electrical conductivities. As such, the larger size of the clusters gives rise to the lower percolation transition in the non-conductive matrices, which is completely analogous to the polydispersity effect.37 We subsequently carry out a supplementary visual characterization on the nanocomposites having the corresponding GNP loading fractions to observe modifications in the volumetric morphologies. In Fig. S4,† we show micro computed tomography (micro-CT) 3D reconstructed scanning images of the ATSP-GNP foam morphology nanocomposites. For GNP 1 μm, the intrinsic porous morphology does not change with respect to the formation of the percolating network. On the other hand, GNPs of 5 μm and 25 μm demonstrate notable changes in their morphologies obtaining nearly densified structural forms above the percolation transitions (see Table S2† for calculated densities of the nanocomposites).
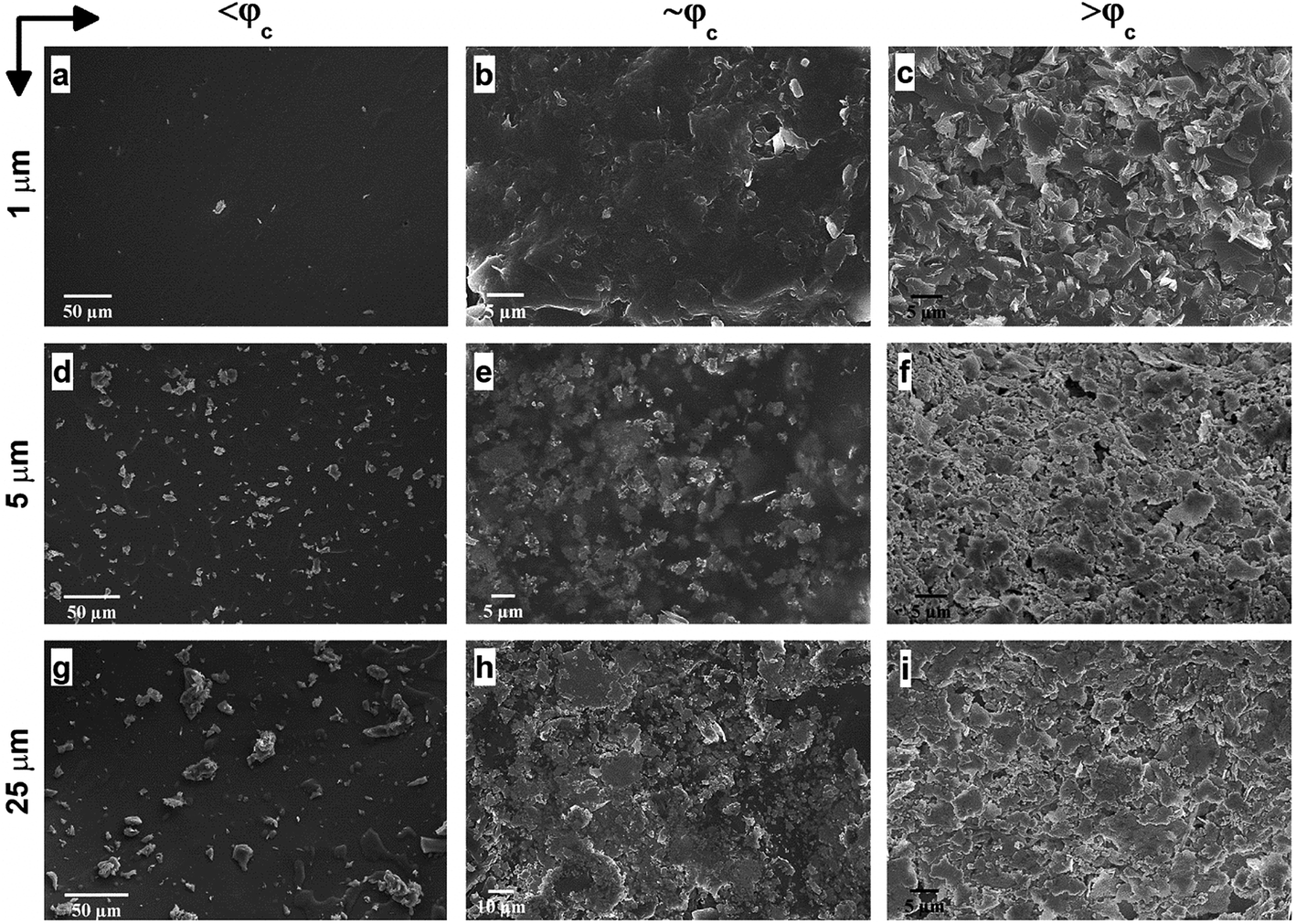 | ||
| Fig. 4 Scanning electron microscopy (SEM) images of the ATSP-GNP nanocomposites highlighting differences in surface morphologies and nanoparticle distributions for the GNPs of 1 μm (a–c), 5 μm (d–f) and 25 μm (g–i) below percolation (<φc), around percolation (∼φc), and above percolation (>φc) thresholds. For GNP 1 μm, <φc, ∼φc, >φc correspond to 1 wt%, 5 wt%, and 10 wt%, respectively. For GNP 5 μm, <φc, ∼φc, >φc correspond to 1 wt%, 4 wt%, and 10 wt%, respectively. For GNP 25 μm, <φc, ∼φc, >φc correspond to 1 wt%, 3 wt%, and 10 wt%, respectively. | ||
3.3. Thermal cure characteristics and backbone chain modifications
To investigate the chemical basis that causes the structural modifications with the increased GNP contents, we analyze the thermal characteristics of the endothermic condensation polymerization reaction at the percolation-determined loading levels. The combined mixtures of the precursor oligomers with the GNPs are subjected to a temperature-ramp differential scanning calorimetry (DSC) cycle at a 10 °C min−1 constant heating rate. The obtained thermal profiles of the GNP combined mixtures are then compared to that of the neat mixture of the constituent oligomers.25 In Fig. 5, we compare only the above-percolation GNP content to the parent material to highlight the thermochemical changes that are effective for other GNP sizes. For comprehensive results of all the GNP sizes at given percolation-determined loading levels, please refer to ESI (Fig. S5–S7†). As shown in Fig. 5, at the beginning of the heating processes, the heat flux curve of the GNP combined mixture likewise reveals a dimple around 70 °C denoting structural softening of the constituent oligomers, which is similarly observed in the base formulation.25 Afterwards, especially for GNP 1 μm, the thermal profile maintains similarly endothermic patterns akin to the neat mixture during which melting and polycondensation reaction stages accordingly take place (Fig. S5†). However, both GNP 5 μm and GNP 25 μm reveal significant heat uptakes into the molten oligomer domain, which are particularly observed for the above-percolation loading levels (>φc) (Fig. S6 and S7†). Such abrupt behaviors are related to acetic acid discharge-driven bubble growth during the condensation reaction that is greatly affected by the presence of the carbon fillers.38 Following this stage, the cure process initiates (denoted as cure onset) around at 260 °C for the parent form in which the thermal curve displays a genuine endothermic cure region.25 For GNP 1 μm, only the >φc loading level exhibits a distinctly broader cure region than the neat oligomers for which the completion of the cure process extends beyond 350 °C (Fig. S5†). In addition, upon completion of the cure process, a thermal relaxation peak, a small hump (denoted as cure end, indicated with an arrow) is observed. Likewise, for both GNP 5 μm and GNP 25 μm, similar broadening of the cure domains as well as the peak formations are observed at above-percolation loading levels. The broadening of the cure region is likely to occur due to increased melt viscosity of the oligomers caused by the presence of the nanofillers that required more energy input to carry out the crosslinking process, which could subsequently stimulate an early gelation of the crosslinked matrix.39 Also, the reactive functional groups of the oligomers graft onto carbon nanofillers that can alter viscous characteristics of the molten domain during curing.40 The thermal relaxation peaks form due to devitrification of the matrix in the thermal process during which the glass transition temperature increased above the cure temperature.41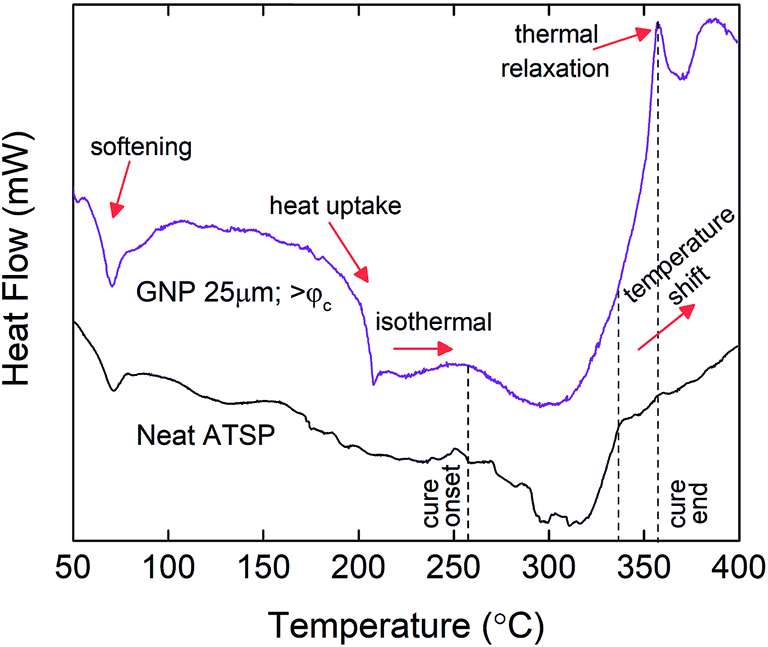 | ||
| Fig. 5 Differential scanning calorimetry (DSC) thermal characteristics of the condensation polymerization reaction for the neat mixture of the precursor oligomers and combined mixture of the oligomers with GNP 25 μm having >φc loading level (10 wt%). DSC curves are arbitrarily shifted to highlight differences. For comprehensive results of all the GNP sizes at given percolation-determined loading levels, please refer to ESI (Fig. S4†). Tests are performed under an inert atmosphere of nitrogen. The heating rate is 10 °C min−1. | ||
In addition, in Fig. 6, we demonstrate solid-state Nuclear Magnetic Resonance (ssNMR) spectroscopy measurements performed on above-percolation content of GNP 25 μm in comparison to the parent material to identify chemical modifications in the backbone chain configuration as well as manifesting the interfacial attachment scheme in the nanofiller incorporated matrix. For comprehensive results of all the GNP sizes at given percolation-determined loading levels, refer to ESI (Fig. S8–S10†). 13C cross-polarization magic-angle spinning (CPMAS) spectrum of the parent material (neat ATSP) display the two characteristic peak domains: the aromatic groups (C–C and C–H) and the functional side groups (C–O and CO), with a line width of ∼860 Hz over the highest intensity peak (∼130 ppm) (13C NMR spectra are processed using 25 Hz line broadening). 1H direct pulse magic-angle spinning (DPMAS) NMR spectrum of the neat ATSP shows a single characteristic having a line width of ∼1640 Hz (1H NMR spectra are processed using 1 Hz line broadening). Based on these NMR analyses, we observe that both 1H and 13C spectra of the nanocomposites with GNPs 5 μm (Fig. S9†) and 25 μm (Fig. 6 and S10†) reveal clearly broadened characteristic peaks with respect to the neat parent matrix. For 1H, GNP 5 μm demonstrates a line width of 2257 Hz at percolation content in comparison to 1640 Hz of the neat matrix. Similarly, for 13C, GNP 5 μm displays a line width of 1528 Hz at percolation content with respect to 860 Hz of the neat ATSP. Whereas GNP 1 μm display characteristic peaks similar to the base ATSP, yet decreased line widths with respect to increased GNP content may indicate only moderate interactions with the matrix (Fig. S8†). For GNPs 5 μm and 25 μm, such peak broadening is formed due to strong interfacial coupling between the GNP particles and ATSP backbone chain, which then modifies the structural relaxation behavior of the nanocomposite matrix.42,43 In particular, the attachment scheme incurs an electron mobility difference due to individually dissimilar relaxation times of highly conductive GNP particles and highly dielectric ATSP matrix. More importantly, we do not observe any characteristic peaks in 13C spectra coming from the matrix of GNP 5 μm at above-percolation loading level (Fig. S9†). As well, 13C spectrum of the GNP 25 μm for >φc form a single slightly broader peak associated with only the aromatic group of the backbone configuration (Fig. 6 and S10†). Hence, the presence of larger GNP particles alter polymerization process at a significant extent, as also justified with the above-mentioned results.
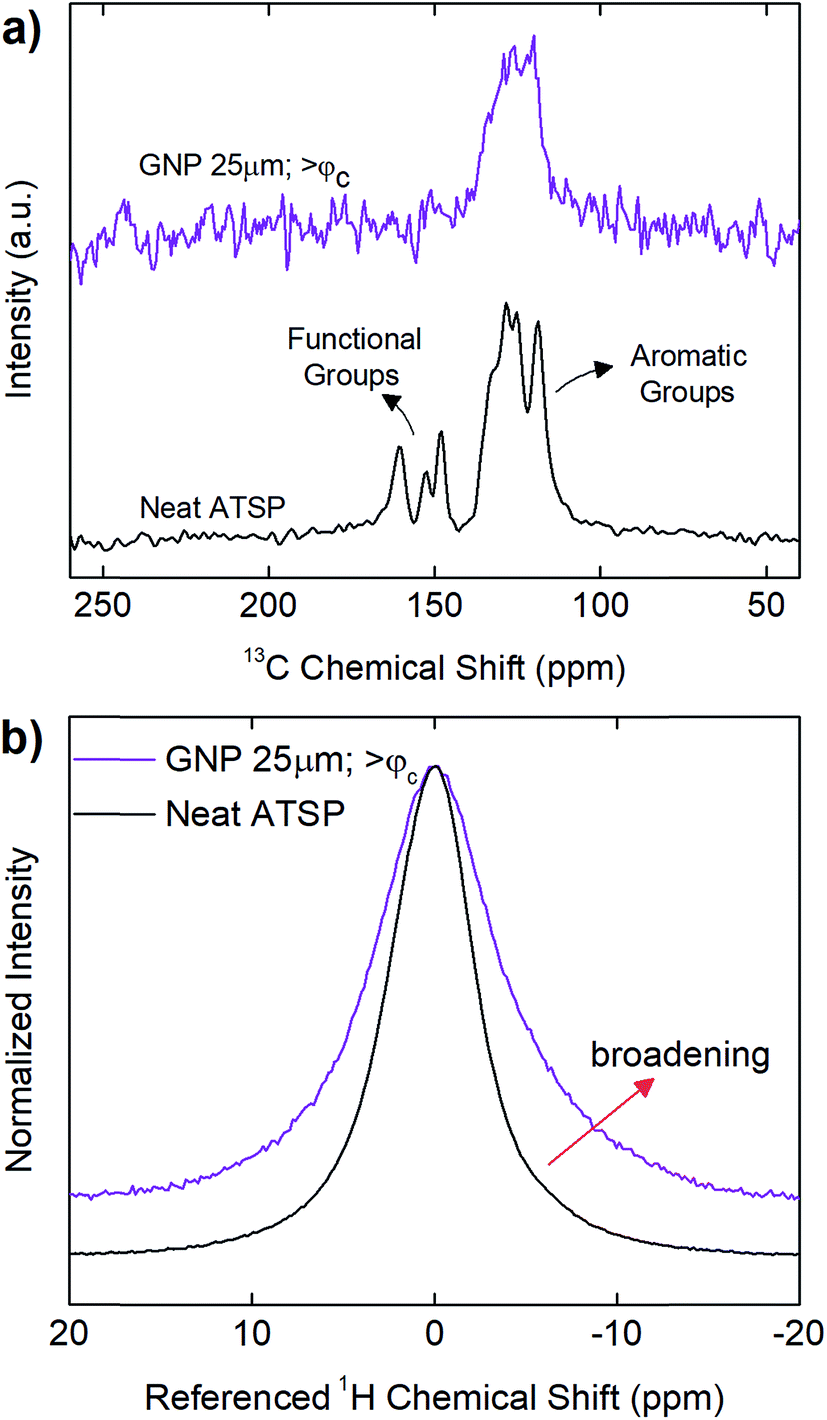 | ||
| Fig. 6 Solid-state nuclear magnetic resonance (ssNMR) 13C cross-polarization magic-angle spinning (CPMAS) (a) and 1H direct pulse magic-angle spinning (DPMAS) (b) spectra of the neat ATSP morphology with respect to ATSP nanocomposites with GNP 25 μm having >φc loading level (10 wt%). 1H and 13C NMR spectra are processed using 1 Hz and 25 Hz line broadenings, respectively. The figures were given in normalized intensity axes. | ||
Based on the characterization results presented here, we understand that due to the interfacial attachment mechanism coming into effect during the polymerization reaction, a fraction of the functional groups of the oligomer structures are reacted with the GNP particles (especially 5 μm and 25 μm), which consequently reduces the fraction of the functional groups that participate in the ordinary crosslinking polymerization process. Indeed, the GNP-coupled configurations of the oligomers prompt an early gelation point during the polymerization reaction without necessitating completion of the crosslinking process. As a consequence of the gelation, the DSC profiles of 5 μm and 25 μm with loadings near and above their percolation thresholds display nearly flat regions following the sudden heat uptakes until cure onset. Under such a circumstance, the modified polymerization process does not solely perform the esterification reaction, which conjugates the two oligomer groups releasing acetic acid by product. This reduces the foaming agent released during the cure process, rendering higher density morphologies as observed in GNP 5 μm and 25 μm above percolation transitions. In this context, the temperature-wise broadening characteristic observed in the cure regions indicates that the advancing crosslinking resin requires more heat input to activate unreacted bonds to complete the cure process. Due to the effect of such an interfacial entanglement mechanism, glass transition characteristics of similar polymer nanocomposites are demonstrated to be modified displaying significant temperature shifts.13 Hence, in this case, the glass transition temperature (∼191 °C) may positively shift to a higher temperature in the cure region such that the polymer chains display the glass transition relaxation while the isothermal cure process is still in progress.25 Therefore, the peak formation at cure end point takes place due to devitrification (phase transformation from glassy state to rubbery state upon the glass transition) of the polymer network. Note that the vitrification induces slower cure rates wherein the cure reaction becomes more diffusion controlled rather than kinetic controlled, which also correlates to the cure region temperature broadening.44 Characterization of rheological properties of the nanofiller incorporated ATSP resin during the in situ polymerization reaction will be the subject of a follow-up study.
4. Conclusions
We present the physicochemical characteristic effects influenced by the GNPs of different sizes on the polycondensation reaction of the ATSP resin via the formation of electrically conductive percolating network. During the polymerization process, carboxylic acid and acetoxy-capped precursor oligomers, combined with the GNPs, form the crosslinked aromatic backbone of the ATSP matrix while the nanofillers interact with the reactive caps of the oligomers and combine with the crosslinked polymer network. The varied GNP size causes different electrical percolation thresholds and ultimate electrical conductivities. Microstructural characterization displays the GNP distributions in the matrix as well as morphological modifications formed through the conductive percolating networks. Cure characteristics reveal that the thermochemical changes occurred on the polymerization processes. ssNMR spectroscopies on the ATSP nanocomposite morphology exhibit the formation of a robust interfacial coupling mechanism between the GNPs and ATSP backbone.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge characterization instruments provided at Frederick Seitz Materials Research Laboratory, Mechanical Testing Instructional Laboratory, Nuclear Magnetic Resonance/Electron Paramagnetic Resonance Laboratory, and Beckman Institute for Advanced Science and Technology at the University of Illinois at Urbana-Champaign, and thank research staff members for their help with the experiments. We sincerely thank Prof. Alexey Bezryadin and Prof. Eduard Ilin for the help with the electrical conductivity measurements. We gratefully acknowledge funding from the National Science Foundation (NSF) I/UCRC grant (IIP-1362146). The findings, conclusions, and recommendations expressed in this manuscript are those of the authors and do not necessarily reflect the views of the NSF.References
- J. C. Williams and E. A. Starke, Acta Mater., 2003, 51, 5775–5799 CrossRef CAS.
- T. M. Pollock, Science, 2010, 328, 986–987 CrossRef CAS PubMed.
- J. Hirsch, Mater. Trans., 2011, 52, 818–824 CrossRef CAS.
- M. Telford, Mater. Today, 2004, 7, 36–43 CrossRef CAS.
- M. Bakir, J. L. Meyer, J. Economy and I. Jasiuk, Polymer, 2017, 123, 311–320 CrossRef CAS.
- P. Podsiadlo, A. K. Kaushik, E. M. Arruda, A. M. Waas, B. S. Shim, J. Xu, H. Nandivada, B. G. Pumplin, J. Lahann, A. Ramamoorthy and N. A. Kotov, Science, 2007, 318, 80–83 CrossRef CAS PubMed.
- J. Jordan, K. I. Jacob, R. Tannenbaum, M. A. Sharaf and I. Jasiuk, Mater. Sci. Eng., R, 2005, 393, 1–11 CrossRef.
- M. Moniruzzaman and K. I. Winey, Macromolecules, 2006, 39, 5194–5205 CrossRef CAS.
- V. Yudin, J. U. Otaigbe, V. Svetlichnyi, E. Korytkova, O. Almjasheva and V. Gusarov, eXPRESS Polym. Lett., 2008, 2, 485–493 CrossRef CAS.
- J. Liang, Y. Huang, L. Zhang, Y. Wang, Y. Ma, T. Guo and Y. Chen, Adv. Funct. Mater., 2009, 19, 2297–2302 CrossRef CAS.
- H. Kim, A. A. Abdala and C. W. Macosko, Macromolecules, 2010, 43, 6515–6530 CrossRef CAS.
- J. R. Potts, S. H. Lee, T. M. Alam, J. An, M. D. Stoller, R. D. Piner and R. S. Ruoff, Carbon, 2011, 49, 2615–2623 CrossRef CAS.
- K.-H. Liao, S. Aoyama, A. A. Abdala and C. Macosko, Macromolecules, 2014, 47, 8311–8319 CrossRef CAS.
- H. Wu, G. Yu, L. Pan, N. Liu, M. T. McDowell, Z. Bao and Y. Cui, Nat. Commun., 2013, 4, 1943 Search PubMed.
- G. Shi, M. Rouabhia, Z. Wang, L. H. Dao and Z. Zhang, Biomaterials, 2004, 25, 2477–2488 CrossRef CAS PubMed.
- P. Karimi, M. Ostoja-Starzewski and I. Jasiuk, J. Appl. Phys., 2016, 120, 145103 CrossRef.
- S. Kale, F. A. Sabet, I. Jasiuk and M. Ostoja-Starzewski, J. Appl. Phys., 2015, 118, 154306 CrossRef.
- W. Zheng, S.-C. Wong and H.-J. Sue, Polymer, 2002, 43, 6767–6773 CrossRef CAS.
- A. Shalwan and B. Yousif, Mater. Des., 2014, 59, 264–273 CrossRef CAS.
- W. Bauhofer and J. Z. Kovacs, Compos. Sci. Technol., 2009, 69, 1486–1498 CrossRef CAS.
- Z. Spitalsky, D. Tasis, K. Papagelis and C. Galiotis, Prog. Polym. Sci., 2010, 35, 357–401 CrossRef CAS.
- P. Pötschke, T. Fornes and D. Paul, Polymer, 2002, 43, 3247–3255 CrossRef.
- D. Frich, K. Goranov, L. Schneggenburger and J. Economy, Macromolecules, 1996, 29, 7734–7739 CrossRef CAS.
- B. Vaezian, J. L. Meyer and J. Economy, Polym. Adv. Technol., 2016, 27, 1006–1013 CrossRef CAS.
- M. Bakir, J. L. Meyer, J. Economy and I. Jasiuk, Macromolecules, 2016, 49, 6489–6496 CrossRef CAS.
- P. A. Couperus, A. D. H. Clague and J. P. C. M. van Dongen, Org. Magn. Reson., 1978, 11, 590–597 CrossRef CAS.
- W. Bubb, C. Fallick and S. Sternhell, Magn. Reson. Chem., 1977, 9, 167–174 CrossRef CAS.
- F. Wang, L. T. Drzal, Y. Qin and Z. Huang, J. Mater. Sci., 2015, 50, 1082–1093 CrossRef CAS.
- J. Xiang and L. T. Drzal, Polymer, 2012, 53, 4202–4210 CrossRef CAS.
- J. A. King, F. A. Morrison, J. M. Keith, M. G. Miller, R. C. Smith, M. Cruz, A. M. Neuhalfen and R. L. Barton, J. Appl. Polym. Sci., 2006, 101, 2680–2688 CrossRef CAS.
- J. B. Bai and A. Allaoui, Composites, Part A, 2003, 34, 689–694 CrossRef.
- J. T. Choi, D. H. Kim, K. S. Ryu, H.-i. Lee, H. M. Jeong, C. M. Shin, J. H. Kim and B. K. Kim, Macromol. Res., 2011, 19, 809–814 CrossRef CAS.
- S. Kale, F. A. Sabet, I. Jasiuk and M. Ostoja-Starzewski, J. Appl. Phys., 2016, 120, 045105 CrossRef.
- A. Aharony and D. Stauffer, Introduction to percolation theory, Taylor & Francis, 2003 Search PubMed.
- R. H. J. Otten and P. v. d. Schoot, J. Chem. Phys., 2011, 134, 094902 CrossRef PubMed.
- K. Nagata, H. Iwabuki and H. Nigo, Compos. Interfaces, 1998, 6, 483–495 CrossRef.
- A. V. Kyrylyuk and P. van der Schoot, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8221–8226 CrossRef CAS PubMed.
- P. Pötschke, T. D. Fornes and D. R. Paul, Polymer, 2002, 43, 3247–3255 CrossRef.
- C.-Y. M. Tung and P. J. Dynes, J. Appl. Polym. Sci., 1982, 27, 569–574 CrossRef CAS.
- S. Li, M. Xiao, D. Wei, H. Xiao, F. Hu and A. Zheng, Polymer, 2009, 50, 6121–6128 CrossRef CAS.
- I. Fraga, S. Montserrat and J. M. Hutchinson, Macromol. Chem. Phys., 2008, 209, 2003–2011 CrossRef CAS.
- J. Chen, H. Liu, W. A. Weimer, M. D. Halls, D. H. Waldeck and G. C. Walker, J. Am. Chem. Soc., 2002, 124, 9034–9035 CrossRef CAS PubMed.
- A. D. Bain, D. R. Eaton, A. E. Hamielec, M. Mlekuz and B. G. Sayer, Macromolecules, 1989, 22, 3561–3564 CrossRef CAS.
- M. Kamal and S. Sourour, Polym. Eng. Sci., 1973, 13, 59–64 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra12506b |
| This journal is © The Royal Society of Chemistry 2018 |