
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRemediation of trichloroethene (TCE)-contaminated groundwater by persulfate oxidation: a field-scale study
Yu-Chen Changa,
Ting-Yu Chenb,
Yung-Pin Tsaia and
Ku-Fan Chen *a
*a
aDepartment of Civil Engineering, National Chi Nan University, 1 Univ. Rd., Puli, Nantou 54561, Taiwan. E-mail: kfchen@ncnu.edu.tw; Tel: +886-49-2910960 ext. 4983
bDepartment of Landscape Architecture, National Chin-Yi University of Technology, Taiping, Taichung 41170, Taiwan
First published on 10th January 2018
Abstract
This study uses a trichloroethene (TCE)-contaminated site to determine the efficacy of persulfate oxidation for the treatment of TCE-contaminated groundwater. The main objectives of this study are: (1) to evaluate the efficacy of TCE treatment using persulfate with different injection strategies; (2) to determine the persistence of persulfate in the aquifer; (3) to determine the radius of influence (ROI) and transport distance of persulfate and (4) to determine the impact of persulfate on indigenous microorganisms during remediation. TCE concentrations are 0.26 mg L−1 in P143 and 0.361 mg L−1 in P146 and the microbial numbers are 6.1 × 103 CFU mL−1 in P143 and 4.4 × 104 CFU mL−1 in P146, before persulfate is injected. The results of the pilot study show that persulfate eliminates TCE. 100% of TCE is removed in P146 and 95% in P143. Single injection of a total amount of 275 kg of 5% persulfate produces better TCE removal than two half persulfate injections in sequence. The transport distance of persulfate ranges from 3.6 to 4.5 m. Persulfate also persists for 14 days in the aquifer. After persulfate is injected, the total bacterial counts decrease slightly to 2.4 × 103 CFU mL−1 in P143 and 1.8 × 103 CFU mL−1 in P146. When persulfate is consumed, the total bacterial counts increase but there is no recovery of the microbial community. The results show that sequential injections of a large amount of persulfate are suggested to maintain good long-term performance for TCE treatment.
1. Introduction
Soil and groundwater that are contaminated by chlorinated organic compounds have become a widespread problem. Chlorinated solvents, such as trichloroethene (TCE), are mainly used for industrial production, degreasing and dry cleaning and are released into the environment because of leakage from storage tanks and the use of improper disposal procedures.1,2 Since TCE is a possible human carcinogen and has acute toxicity to humans, the contamination of groundwater with TCE is of concern. The US Environmental Protection Agency (EPA) has set a maximum contaminant level (MCL) of 5 μg L−1 for drinking water to protect public health.3In situ chemical oxidation (ISCO) is a widely used technology for the remediation of contaminated soil and groundwater. Conventional ISCO oxidants that are used for the treatment of contaminated sites include hydrogen peroxide (H2O2) solutions, such as Fenton's reagent and Fenton-like reaction, ozone (O3), and permanganate (MnO4−).4 Recently, persulfate has been applied to remediate soil and groundwater contamination because of its high redox potential (reaction (1)).
| S2O82− + 2e− → 2SO42−, E0 = 2.01 V | (1) |
Persulfate is activated by activators such as heat or ferrous ions to generate powerful sulfate free radicals (SO4−˙) (reaction (2)):1,5
| S2O82− + activators → 2SO4−˙ or SO4−˙ + SO42−, E0 = 2.7 V | (2) |
Persulfate oxidation is used to degrade various contaminants, such as chlorinated organic compounds and petroleum hydrocarbons.6,7 Because persulfate is a stable oxidant that persists in the subsurface for a long time, it is a useful oxidant for soil and groundwater remediation.1,8,9 Many studies of persulfate oxidation focus on the development of activation methods,2,10–12 the feasibility of contaminant removal,5,6 the effect of pH on pollutant degradation13–15 and the mechanism and kinetics of contaminant removal.12,16,17 Studies involving the application of persulfate are fewer. Although the results of some field studies and site remediation that use persulfate oxidation have been published, these reports mainly focus on the efficiency with which contaminants are removed.18 However, other practical parameters and information, such as injection strategies, the radius of influence (ROI), the transport distance, the persistence of persulfate, persulfate dosage and the damage that persulfate does to indigenous microorganisms are not widely studied in the field.
This study uses a TCE-contaminated site for a field-scale study, to collect practical information about in situ persulfate oxidation. The main objectives of this study are to determine: (1) the efficiency of TCE treatment using persulfate oxidation; (2) the persistence of persulfate in the aquifer; (3) the ROI and transport distance of persulfate and (4) the impact of persulfate on indigenous microorganisms during remediation.
2. Materials and methods
2.1. Site description
The pilot-scale study was conducted at an industrial park site that is located in southern Taiwan. The area of the industrial park is 978![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 m2. TCE contamination was detected at this site in early 2006. The results of a previous site investigation showed that TCE concentrations in groundwater exceeded the groundwater control standard (GCS) (0.05 mg L−1) that is set by the Taiwanese EPA. The aquifer sediment at this site is loamy sand with a hydraulic conductivity of 0.007 cm s−1. The groundwater flows from southeast to northwest, with an average discharge velocity of 0.5 m per day. The water table is generally at a depth of 5 to 7 m below the ground surface (bgs). A 3 to 5 m thick clay layer is located at 11 m bgs. The properties of the aquifer sediment at the site are shown in Table 1.
000 m2. TCE contamination was detected at this site in early 2006. The results of a previous site investigation showed that TCE concentrations in groundwater exceeded the groundwater control standard (GCS) (0.05 mg L−1) that is set by the Taiwanese EPA. The aquifer sediment at this site is loamy sand with a hydraulic conductivity of 0.007 cm s−1. The groundwater flows from southeast to northwest, with an average discharge velocity of 0.5 m per day. The water table is generally at a depth of 5 to 7 m below the ground surface (bgs). A 3 to 5 m thick clay layer is located at 11 m bgs. The properties of the aquifer sediment at the site are shown in Table 1.
| Item | Measured value |
|---|---|
| pH | 8.42 |
| ORP (mV) | 188 |
| Total iron (mg kg−1) | 40100 |
| Soil organic matter (%) | 2.52 |
| Total bacterial count (CFU g−1 soil) | 7.75 × 104 |
| Texture classification | Loamy sand |
| Sand (%) | 85 |
| Silt (%) | 12 |
| Clay (%) | 3 |
A plastic manufacturing factory in the industrial park with an area of 8750 m2 was selected for the field-scale study. An estimated TCE plume of approximately 5200 m2 was present in the subsurface. An area of 16 m × 9 m was selected for the pilot study. Seven 1-inch wells were installed at this site. Fig. 1 shows the site map, showing the groundwater flow direction, two injection wells (P143 and P146), a background well (P147) and four monitoring wells (P142, P144, P145, and TW38) at the test site. All wells were approximately 10 m in depth, with screen intervals from 6 to 10 m bgs.
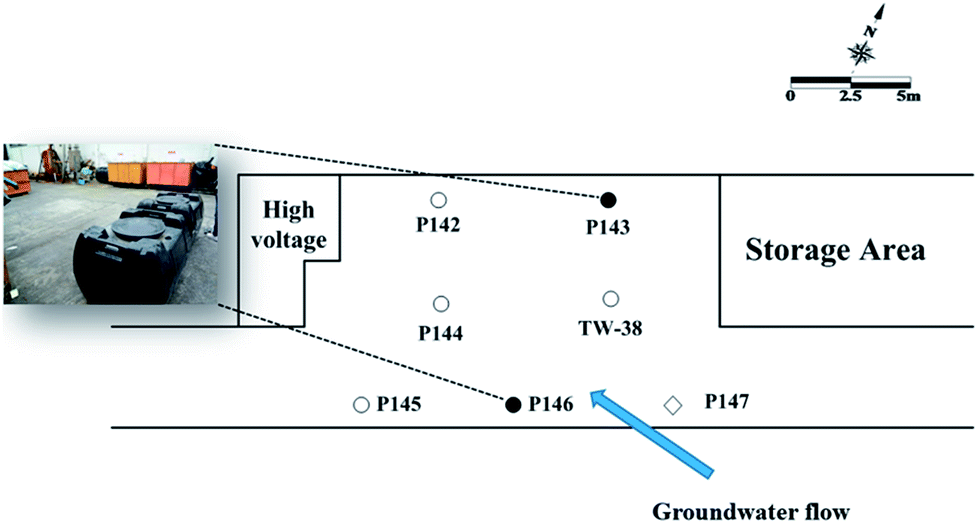 | ||
| Fig. 1 Site map showing the groundwater flow direction, the injection wells, the background well and other monitor wells at the test site (●: injection well; ○: monitor well; ◇: background well). | ||
2.2. Persulfate injection
Persulfate solution (5% (wt/wt)) was prepared in 500 L polyethylene tanks and was then pumped into the injection wells. A single injection of persulfate (275 kg) and a two-stage injection of persulfate (150 kg followed by 125 kg) were respectively used for wells P146 and P143, in order to evaluate the effect of different injection strategies on the removal of TCE and the persistence of persulfate. Each injection well had two 500 L tanks and persulfate was pumped from these two tanks in sequence at an injection flow rate of about 10 L min−1. When one of the tanks was empty, the other tank was then used for persulfate injection and the empty tank was refilled with persulfate solution for the subsequent injection. A total of approximately 5500 L of 5% persulfate solution (275 kg/0.05 = 5500 kg ≈ 5500 L) was injected into each injection well during the field test.2.3. Groundwater sampling and analysis
Groundwater was periodically sampled from all wells, according to US EPA.19 TCE analysis was performed using a gas chromatography-flame ionization detector (GC-FID) (7890A, Agilent Technologies, USA). The GC-FID used a capillary column (GsBP-624, 60 m × 0.32 mm, GS-Tek, USA) with nitrogen gas (99.9995% purity) flowing at 2.4 mL min−1 as the carrying gas. The operating temperature for the injector was maintained at 180 °C and that for the detector at 230 °C. The oven temperature was initially maintained at 35 °C for 5 min and then increased at a rate of 11 °C min−1 to 115 °C and held at 115 °C for 3 min. The temperature was then increased at a rate of 20 °C min−1 to 220 °C and then held at 220 °C for 1 min. The persulfate content was analyzed using a spectrophotometer (DR 5000, Hach, USA) using the method of Huang et al.13 Alkalinity was measured according to APHA.20 Total iron and ferrous iron contents were analyzed using the Hach spectrophotometer using Hach methods 8008 and 8146, adapted from Standard Methods for the Examination of Water and Wastewater, respectively.21 All of the samples were stored at 4 °C before analysis. The values for pH (pH 3210, WTW, Germany), oxidation–reduction potential (ORP) (Multi 3210, WTW, Germany) dissolved oxygen (DO) (Oxi 3210, WTW, Germany), and CO2 were measured in the field. A Hach digital titrator cartridge was used for CO2 measurements (Hach Method 8205).2.4. Microbial analysis
A total bacterial count (TBC) was performed to determine the effect of persulfate on intrinsic bacterial numbers, using a Difco™ plate count agar (Becton, Dickinson and Co., USA) (APHA, 1998).20 The prepared plates were incubated at 30 °C for 48 hours and then counted for colony forming units (CFU). Polymerase chain reaction-denaturing gradient gel electrophoresis (PCR-DGGE) analysis was used to determine the impact of persulfate on the indigenous microbial community. DNA extraction was performed for 0.25 g of soil samples using a PowerSoil® DNA Isolation Kit (Mo Biol, USA). The V6–V8 region of the 16S rDNA was amplified using the primers, 968F (5′-AAC GCG AAG AAC CTT AC-3′) and 1401R (5′-CGG TGT GTA CAA GAC CC-3′). PCR amplification used an initial denaturation at 97 °C for 5 min and then 28 cycles of annealing at 9 °C for 1 min, 54 °C for 40 s and 72 °C for 30 s, followed by a final extension at 72 °C for 7 min. Each amplified PCR product was subjected to DGGE using a Bio-Rad DCode system (Bio-Rad, Hercules, CA, USA), to monitor the changes in microbial diversity during the field test.3. Results and discussion
3.1. Site characterization
Table 2 shows the results for chlorinated ethenes and the geochemical indicators in all wells before persulfate injection. The TCE concentrations in all wells, except for TW-38, exceed the groundwater control standard of 0.05 mg L−1 (4.9 to 7.7 times). 1,1-Dichloroethene (DCE) (0.008 to 0.017 mg L−1) and cis-DCE (0.003 to 0.008 mg L−1) are also present in groundwater. trans-DCE and vinyl chloride (VC) are not detected. The TBC for all wells ranges from 2.8 × 103 to 3.13 × 105 CFU mL−1. Generally, biodegradation is one of the most important mechanisms for the natural attenuation of contaminants. The results for ORP and DO measurement show that the groundwater at this site is aerobic. Although TCE is not removed via direct metabolism under aerobic conditions, it can be biodegraded by aerobic co-metabolism, using soil organics as the primary carbon source. However, co-metabolic biodegradation of TCE under aerobic conditions is limited if no extra carbon source is added.1,22,23 Because TCE contamination was first detected in 2006, the results indicate that natural attenuation mechanisms at this site did not decrease TCE concentrations to the groundwater control standard. Therefore, more active remediation technologies are required for this site. Groundwater alkalinity is between 268 and 315 mg L−1 as CaCO3, which indicates that the groundwater has a buffer capacity for acid. Significant total iron and trace ferrous iron are present in all wells. The existence of iron species might favor the removal of TCE effectively because iron can serve as an activator for the activation of persulfate.4 It should be noted that groundwater with a high buffering capacity can result in low concentrations of soluble iron. In fact, the ferrous iron concentrations are low at this site because of the aerobic conditions and the buffering capacity of the groundwater. The high concentrations of total iron may come from the aquifer sediment particles in groundwater samples because there is a high iron content in the aquifer sediment (Table 1). The collected samples were preserved by adding nitric acid (pH < 2), so iron oxide may be dissolved and released from the particles.| a Injection well.b Monitoring well.c Background well.d Not detectable.e Total bacterial count. | |||||||
|---|---|---|---|---|---|---|---|
| Wells | P142 | P143 | P144 | P145 | P146 | P147 | TW-38 |
| Well function | MWb | IWa | MWb | MWb | IWa | BWc | MWb |
| TCE (mg L−1) | 0.151 | 0.264 | 0.245 | 0.384 | 0.361 | 0.245 | 0.040 |
| 1,1-DCE (mg L−1) | 0.008 | 0.011 | 0.007 | 0.013 | 0.017 | 0.006 | 0.016 |
| cis-DCE (mg L−1) | 0.003 | 0.006 | 0.003 | 0.006 | NDd | NDd | 0.008 |
| trans-DCE (mg L−1) | NDd | NDd | NDd | NDd | NDd | NDd | NDd |
| VC | NDd | NDd | NDd | NDd | NDd | NDd | NDd |
| Temperature (°C) | 31.3 | 30.4 | 31.2 | 31.6 | 30.1 | 30.4 | 30.5 |
| pH | 6.70 | 7.06 | 7.07 | 7.22 | 7.22 | 7.12 | 7.00 |
| ORP (mV) | 275.2 | 250.2 | 321.3 | 265.3 | 261 | 275.1 | 115 |
| DO (mg L−1) | 2.2 | 1.93 | 1.44 | 1.35 | 1.35 | 1.28 | 1.38 |
| Total iron (mg L−1) | 26.5 | 28.9 | 23.2 | 16.5 | 34 | 18.5 | 64.3 |
| Ferrous iron (mg L−1) | 0.220 | 0.355 | 0.065 | 0.345 | 0.050 | 0.105 | 1.605 |
| CO2 (mg L−1) | 250 | 140 | 172 | 76 | 52 | 32 | 183 |
| Alk (mg L−1 as CaCO3) | 341 | 294 | 268 | 312 | 312 | 293 | 315 |
| TBCe (CFU mL−1) | 2.8 × 103 | 6.1 × 103 | 3.13 × 105 | 8.9 × 104 | 4.4 × 104 | 5.1 × 104 | 3.6 × 104 |
3.2. TCE removal by in situ persulfate oxidation
In this study, a single injection of persulfate (275 kg) and a two-stage injection of persulfate (150 kg followed by 125 kg) were applied to P146 and P143, respectively, in order to evaluate the effect of different injection strategies on the removal of TCE. As show in Table 2, since the concentrations of 1,1-DCE and cis-DCE are much lower than that of groundwater control standards (0.070 mg L−1 for 1,1-DCE and 1.0 mg L−1 for cis-DCE), this study only focuses on the discussion of the efficacy of TCE treatment by persulfate. Fig. 2 shows the results of the pilot-scale study. After persulfate is injected, TCE concentrations drop from 0.361 to 0 mg L−1 in P146 and from 0.264 to 0.012 mg L−1 in P143 within 2 days (Fig. 2(a)). TCE removal efficiency is 100% in P146 and 95% in P143. Single injection of a total amount of 275 kg of 5% persulfate produces better TCE removal than two half persulfate injections in sequence.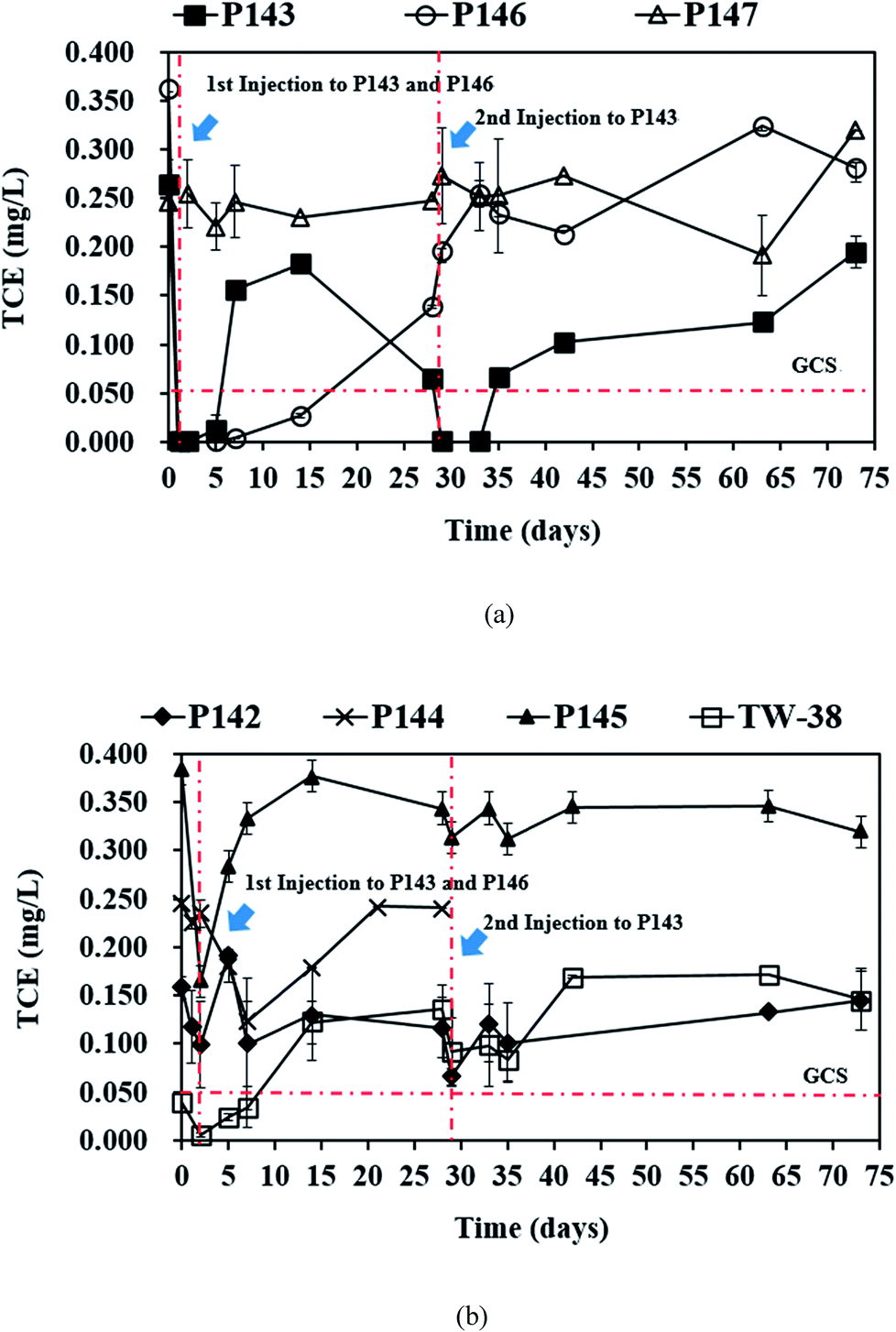 | ||
| Fig. 2 The results of the pilot-scale study: (a) TCE concentrations in P143, P146, and P147 and (b) TCE concentrations in P142, P144, P145 and TW-38 (P146: single injection; P143: two-stage injection; persulfate: 5%). | ||
In a persulfate system, pH is changeable due to the generation of bisulfate (HSO4−) during the decomposition of persulfate.24 Decreases of 2.5 to 6.0 pH units caused by persulfate have been observed in aqueous and soil slurry systems.1 In general, pH plays an important role in affecting the performance for the degradation of contaminants in persulfate systems. Hussain et al.24 found that acid conditions (pH 2.0 and pH 4.0) are favorable to the degradation of p-chloroaniline (PCA) in a zero-valent iron-activated persulfate system. Additionally, PCA degradation at pH 7 is higher than that at pH 9 and pH 11. Liang et al.14 indicated that maximum TCE degradation occurs at pH 7 in persulfate systems, which are activated by ambient temperatures (10 to 30 °C). It has also been demonstrated that high pH conditions may cause a decrease in the oxidation of contaminants.15,24 Therefore, acid and neutral conditions seem to be advantageous to the degradation of pollutants in persulfate systems according to the above reports. In this study, pH decreases to approximately 5.80 after persulfate is injected and then pH in the injection wells remains near neutral because of the buffering capacity of groundwater. The results show that pH conditions at this site are favorable to the degradation of TCE during the field study.
Liang et al.14 and Fang et al.25 noted that sulfate radicals are produced at ambient temperature (10–30 °C) by thermal activation, which are predominant under acid and neutral conditions. In this study, the temperature and pH in groundwater are between 30.4 to 31.6 °C and 6.70 to 7.22, respectively (Table 2). pH slightly decreases to around 5.80 after persulfate is injected to the wells and then increases to above 6.00. This indicates that sulfate radicals are dominant, which contribute the removal of TCE during the field test. Waldemer et al.26 also reported that the rate-determining step for chlorinated ethane oxidation by heat activated persulfate involved the reaction with sulfate radicals but not the formation of sulfate radicals. In addition, sulfate radicals can react with water to produce hydroxyl radicals under all pH conditions, as shown in reaction (3).26 Thus, hydroxyl radicals may also be responsible for the degradation of TCE in groundwater.
| SO4˙− + H2O → SO42− + ˙OH + H+ | (3) |
Although it has been reported that persulfate might be activated by native soil minerals, such as iron to enhance contaminant degradation rates in a slurry system,27 more recent studies indicated that major subsurface minerals do not activate persulfate.28,29 Ahmad et al.28 found that synthetic iron and manganese oxides are able to activate persulfate. However, the masses of iron and manganese oxides typically found in natural soil are not sufficient to activate persulfate. TCE oxidation by sulfate free radicals can be simply described as reaction (4):14
| SO4˙− + C2HCl3 + 4H2O → 2CO2 + 9H+ + 3Cl− + 6SO42− | (4) |
Yan et al.30 indicated that hydroxyl radicals converted TCE to dichloroacetic acid via the electrophilic addition reaction of TCE:
| ˙OH + C2HCl3 → CCl2CHOOH + Cl− | (5) |
According to the above discussion, sulfate radicals produced by thermal activation and hydroxyl radicals derived from sulfate radicals play a major role in the destruction of TCE during the field test.
Oxidation intermediates of TCE are not identified during remediation. Although the issue of determining the pathway of TCE degradation by persulfate has been mentioned, the information regarding intermediates of TCE oxidation by persulfate is limited.14 Yan et al.30 recently reported that formic acid and dichloroacetic acid were detected during TCE oxidation by siderite-catalyzed hydrogen peroxide coupled with persulfate. Therefore, more studies on this issue are needed in the future.
The injection of persulfate into the groundwater may reduce the concentrations of pollutants via dilution.4,11 It should be noted that CO2 concentrations increase from the background level of 140 mg L−1 to 835 mg L−1 in P143 and from 52 mg L−1 to 584 mg L−1 in P146 after 2 days of persulfate injection. Although it is difficult to distinguish the effect of oxidation and dilution on the decrease in TCE concentrations by persulfate, the increase in CO2 concentrations in the injection wells provide strong evidence that an extent of TCE and aquifer organics are certainly mineralized by the injected persulfate. Additionally, the TCE that is adsorbed onto the sediment may be a source for the continuous release of TCE to the groundwater. The injected persulfate treats the soluble TCE in the groundwater and degrades the adsorbed TCE in the aquifer sediment. Contaminants in soil and groundwater are effectively removed by persulfate,11,30 so although TCE in groundwater may be diluted by the injected persulfate, persulfate still contributes to the removal of TCE in both aqueous and soil-sorbed phases. Groundwater flow may also affect the elimination of TCE. Although TCE in the pilot study area may be removed by groundwater flow, TCE is transported to the treatment zone from the upgradient area continuously. Since oxidation also contributes TCE degradation, it is difficult to determine the contribution of hydraulic dynamics to the removal of TCE at this site.
The single injection process maintains the concentration of TCE in P146 at less than 0.05 mg L−1 for 14 days. However, the TCE concentration increases to 0.138 mg L−1 on day 28. The TCE concentration in P143 is reduced only for 5 days and then rapidly increases to 0.153 mg L−1 on day 7. These results show that the injection of a large amount of persulfate results in more stable TCE remediation. When the second injection is applied to P143 on day 28, TCE concentration decreases immediately and then increases to more than the groundwater control standard after 7 days of reaction. The TCE concentrations in P144 and P145, the downgradient monitoring wells of P146, decrease significantly at day 2 and then increase again at day 7 (Fig. 2(b)). Groundwater was not sampled in P144 after day 28 because a bailer was accidently dropped into the well, causing a blockage of the well. In TW-38, a decrease in TCE concentrations is observed after 2 days and then the concentrations of TCE increase with time. The decrease in the TCE concentrations in P144, P145, and TW-38 may be due to persulfate seepage from P146 (Fig. 3(a)). Although there is a slight decrease in the TCE concentrations in P142, which is the downgradient monitoring well of P143, during the first 2 days of persulfate injection, the variation may be caused by the heterogeneity of the subsurface, because no persulfate is detected in P142. The TCE concentration in the background well P147 is stable during the field study.
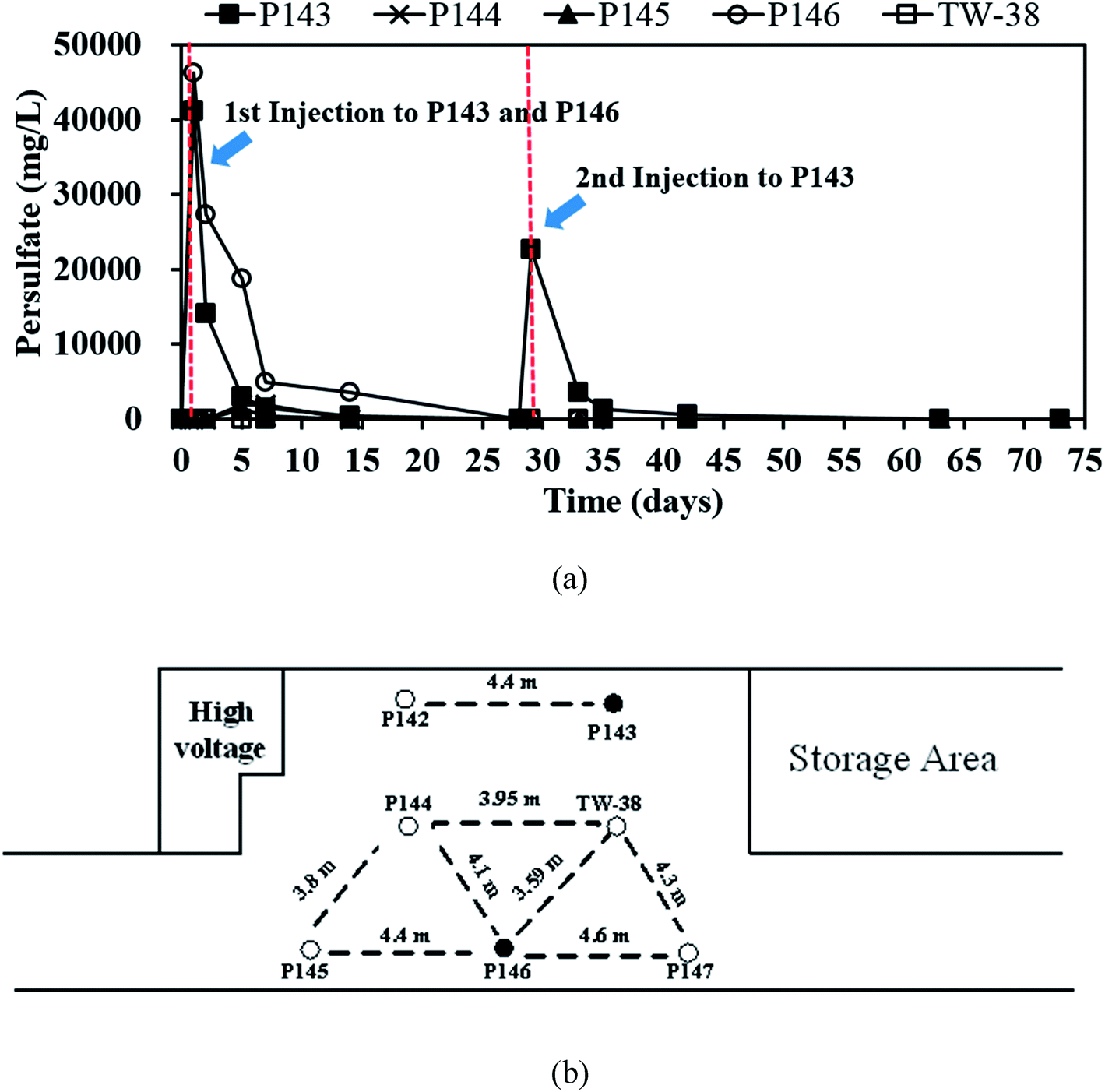 | ||
| Fig. 3 (a) The variation in persulfate concentrations in P143 to P146 and TW-38 and (b) the distance between each well. | ||
Fig. 3(a) shows that on day 14, persulfate concentration in P146 decreases from an initial value of about 46000 to 3500 mg L−1 and the TCE concentration is still less than 0.05 mg L−1 (Fig. 2(a)). This indicates that the presence of persulfate inhibits any further increase in TCE. Similar results are also observed for P143 on day 5. Do et al.31 reported that sequential injections of peroxymonosulfate (PMS)/Co(II) improves the degradation of diesel in soils. When the concentration of PMS increases, fewer injections are needed to achieve remediation. The results of this pilot study suggest that sequential injections of a large amount of persulfate allow good long-term TCE treatment. The TCE concentrations increase to approximately 0.300 mg L−1 in P146 and 0.200 mg L−1 in P143 at the end of the experiment (Fig. 2(a)), probably because TCE is transported from the upgradient areas.
3.3. The persistence of persulfate in the aquifer
Fig. 3(a) shows that after persulfate is injected, persulfate concentrations are 41210 mg L−1 in P143 and 46260 mg L−1 in P146. These values respectively decrease to 14110 and 27310 after 2 days of reaction. Persulfate persists in the aquifer for 14 days in P146 but the persulfate in P143 is almost completely depleted after 7 days of injection. The results show that persulfate is more persistent in an aquifer where there is a large amount of injection. In laboratory studies, Sra et al.8 reported that un-activated persulfate has a half-life of 2 to 600 days in various uncontaminated aquifer materials. Chen et al.9 also showed that 1% of persulfate still exists in a slurry system for at least 80 days. However, persulfate only persists for 14 days in this study, probably because of the movement of groundwater and consumption by the large amount of soil organic matter in the aquifer sediment. Therefore, when persulfate is applied in the field, it is necessary to monitor persulfate concentrations to ensure effective removal of the contaminant.
Since no persulfate is detected in other monitoring wells after all persulfate is injected to P143 and P146, the ROI for persulfate injection is not determined in this study. Therefore, more monitoring wells closer to the injection wells are needed in the future to obtain the ROI for persulfate delivery. Persulfate is detected in P144 (464 mg L−1), P145 (1292 mg L−1) and TW-38 (98 mg L−1) after 5 days of operation. The distance between each well at this site (Fig. 3(b)) gives an estimated value for the transport distance of persulfate of 3.6 to 4.5 m. The transport distance of persulfate depends on the rate of decomposition and the velocity. Tsitonaki et al.18 found that transport distances of persulfate ranged from 0.1 to 23.0 m with persulfate decomposition rates between 0.01 to 2.39 1 per day at a seepage velocity of 0.1 m per day. However, the values are based on very limited data. In this study, the transport distance of persulfate was 3.6 to 4.5 m at a groundwater velocity of 0.5 m per day. The transport distance of persulfate obtained from this field test provides additional practical information for in situ persulfate oxidation. The results for pH monitoring show that the groundwater in each injection well remains near neutral pH during the field test (Fig. 4). Therefore, the presence of bicarbonate does not significantly elevate the concentration of CO2.
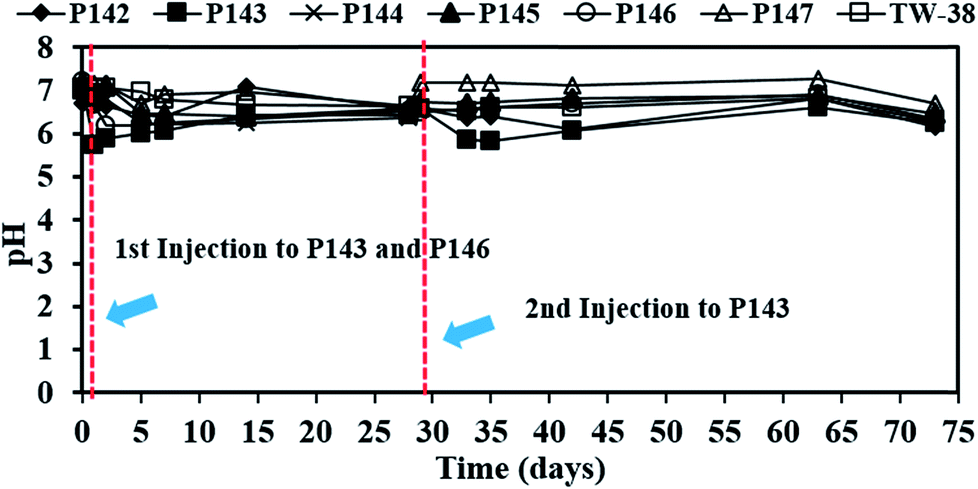 | ||
| Fig. 4 The pH values that were measured for each well during the test. | ||
3.4. The effect of oxidants on indigenous microorganisms
Fig. 5 shows the effect of persulfate on the number of indigenous microorganisms. The numbers of bacteria are 8.1 × 103 CFU mL−1 in P143, 8.9 × 104 CFU mL−1 in P145, and 4.6 × 104 CFU mL−1 in P146, before persulfate is injected. After persulfate injection, the microbial numbers in P143 and P146 slightly respectively decrease to 2.4 × 103 and 1.8 × 103 CFU mL−1 within 2 days and decrease further over the next 7 days of reaction. Microbial numbers increase in both wells and persulfate concentrations in P143 (41200 mg L−1) and P146 (46260 mg L−1) respectively drop to 500 and 3583 mg L−1 on day 14. The number of bacteria in P146 recovers on day 28 and remains at more than 4.5 × 104 CFU mL−1 until the end of the experiment. When the second injection of persulfate is applied to P143 on day 28, the total bacteria count decreases again in this well and then recovers at the end of the test. Bacteria in P145 are also damaged after persulfate moves to the well on day 5 (Fig. 3(a)). However, there is a growth of bacteria after persulfate is depleted. The results show that persulfate does not cause significant damage to the number of indigenous microbes. Recent bench-scale studies have demonstrated that persulfate cause more significant damage to indigenous microorganisms than hydrogen peroxide and permanganate because of its low pH.9,32 The high buffering capacity of the groundwater at this site plays an important role in the protection of in situ microbes.
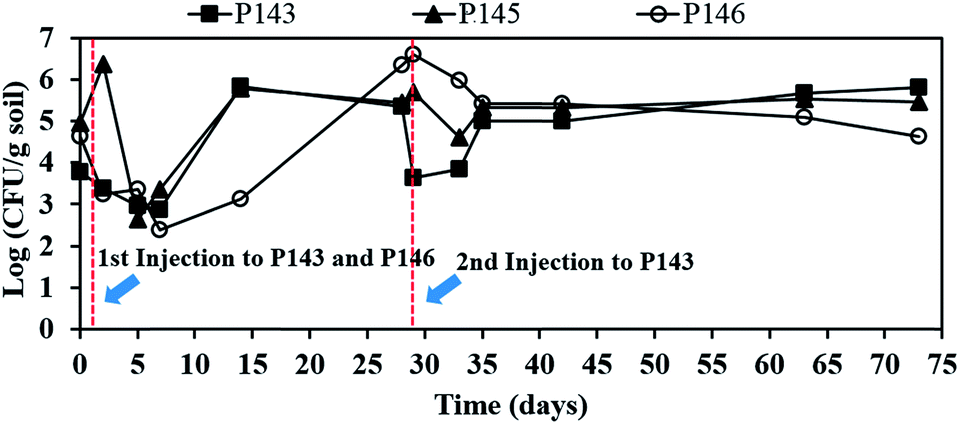 | ||
| Fig. 5 The effect of persulfate on the number of indigenous microorganisms. | ||
Oxidants can change the community and the predominant species of in situ microorganisms, so DGGE analysis was used to monitor the variation in the major microbes during persulfate oxidation. Fig. 6 shows the DGGE profiles for the PCR-amplified 16S rDNA during the pilot-scale study. The green bars signify bacteria that appear in the background before persulfate injection. The yellow bars signify bacteria that do not appear in the background. The red bars signify bacteria that match the bacteria in the background. In well P146, the microbial community changes significantly and the microbial diversity decreases after persulfate is injected. These results are in agreement with the results of other studies. Richardson et al.32 reported that microbial diversity decreases after exposure to persulfate, for a column study. The recovery of specific contaminant degraders also takes longer. Chen et al.9 showed that persulfate causes more significant destruction in a microbial community than hydrogen peroxide or permanganate. It should be noted that although the microbial diversity for this study is still low on day 28 (Fig. (6)), the number of bacteria in P146 recovers (Fig. 5). The results show that only some species of microbes that are resistant to persulfate contribute to the total number of bacteria. In P144, the downgradient well of P146, the microbial diversity also decreases during 5 days of operation. However, the diversity of the microbial community recovers at the end of the test. During the pilot study, low concentrations of persulfate (464 mg L−1) are observed in P144 on Day 5 because there is transport from P146. Although low concentrations of persulfate also have an adverse effect on the microbial community, the damage to the indigenous microbes is not significant.
 | ||
| Fig. 6 DGGE profiles for the PCR – amplified 16 rDNA for groundwater samples (P146: injection well; P144: monitoring well; lane 1: day 0; lane 2: day 2; lane 3: day 5; lane 4: day 7; lane 5: day 14; lane 6: day 21; lane 7: day 28). | ||
4. Conclusions
TCE-contaminated groundwater is successfully treated using in situ persulfate oxidation in this pilot-scale study. The conclusions of this study are as follows:(1) TCE is removed effectively by the injection of 5% persulfate.
(2) Persulfate persists for 14 days in the aquifer. The transport distances for persulfate injection at this site range from 3.6 to 4.5 m.
(3) It is necessary to monitor persulfate concentrations to ensure effective removal of the contaminant.
(4) Although the numbers of total bacterial counts recover after persulfate is depleted, there is no recovery of the microbial community in the injection well P146. Therefore, only some species of microbes that are resistant to persulfate contribute to the total number of bacteria.
(5) Sequential injections of a large amount of persulfate are suggested to give good long-term performance for TCE treatment.
The results of this study will provide useful information for the application of persulfate in the field. More injection protocols and down-gradient wells could be used for this site in the future, to evaluate the effect of the injection processes and ROI for persulfate injection in more detail.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was funded by the Ministry of Science and Technology, Taiwan (103-2622-E-260-002-CC3). The authors would like thank to Mr Tien-Hsing (James) Tung and the personnel of the Apollo Technology Corp., Ltd., Taiwan and graduate students at National Chi Nan University, Taiwan, for their assistance throughout this project.References
- Y. P. Peng, K. F. Chen, W. H. Lin, Y. C. Chang and F. Wu, RSC Adv., 2016, 6, 41247–41260 RSC.
- C. Liang, C. J. Bruell, M. C. Marley and K. L. Sperry, Chemosphere, 2004, 55, 1225–1233 CrossRef CAS PubMed.
- CSWRCB (California State Water Resources Control Board), Groundwater Information Sheet: Trichloroethene (TCE), 2009 Search PubMed.
- ITRC (The Interstate Technology & Regulatory Council), Technical and Regulatory Guidance for In Situ Chemical Oxidation, 2nd edn, 2005 Search PubMed.
- C. Liang, C. J. Bruell, M. C. Marley and K. L. Sperry, Soil Sediment Contam., 2003, 12, 207–228 CrossRef CAS.
- K. C. Huang, Z. Zhao, G. E. Hoag, A. Dahmani and P. A. Block, Chemosphere, 2005, 6, 551–560 CrossRef PubMed.
- S. G. Huling and B. E. Pivetz, In Situ Chemical Oxidation, DTIC Document, 2006 Search PubMed.
- K. S. Sra, N. R. Thomson and J. F. Barjer, Environ. Sci. Technol., 2010, 44, 3098–3104 CrossRef CAS PubMed.
- K. F. Chen, Y. C. Chang and W. T. Chiou, J. Chem. Technol. Biotechnol., 2016, 91, 1877–1888 CrossRef CAS.
- P. A. Block, R. A. Brown and D. Robinson, Novel activation technologies for sodium persulfate in situ chemical oxidation, Proceedings of the 4th International Conference on the Remediation of Chlorinated and Recalcitrant Compounds, 2004 Search PubMed.
- C. Liang, I. L. Lee, I. Y. Hsu, C. P. Liang and Y. L. Lin, Chemosphere, 2008, 70, 426–435 CrossRef CAS PubMed.
- O. S. Furman, A. L. Teel and R. J. Watts, Environ. Sci. Technol., 2010, 44, 6423–6428 CrossRef CAS PubMed.
- K. C. Huang, R. A. Couttenye and G. Hoag, Chemosphere, 2002, 49, 413–420 CrossRef CAS PubMed.
- C. Liang, Z. S. Wang and C. J. Bruell, Chemosphere, 2007, 66, 106–113 CrossRef CAS PubMed.
- K. F. Chen, Y. C. Chang and K. Y. Liu, RSC Adv., 2016, 6, 75578–75587 RSC.
- H. Y. Liang, Y. Q. Zhang, S. B. Huang and I. Hussain, Chem. Eng. J., 2013, 218, 384–391 CrossRef CAS.
- D. Zhao, X. Liao, X. Yan, S. G. Huling, T. Chai and H. Tao, J. Hazard. Mater., 2013, 254–255, 228–235 CrossRef CAS PubMed.
- A. Tsitonaki, B. Petri, M. Crimi, H. Mosbaek, R. L. Siegrist and P. L. Bjerg, Crit. Rev. Environ. Sci. Technol., 2010, 40, 55–91 CrossRef CAS.
- US EPA (Environmental Protection Agency), Ground-Water Sampling Guidelines for Superfund and RCRA Project Managers, EPA/542/S-02/001, 2002 Search PubMed.
- American Public Health Association (APHA), Standard Methods for the Examination of Water and Wastewater, 20th edn, APHA-AWWA-WEF, Washington, DC, 1998 Search PubMed.
- American Public Health Association (APHA), Standard Methods for the Examination of Water and Wastewater, 15th edn, APHA-AWWA-WEF, Washington, DC, 1980 Search PubMed.
- K. F. Chen, T. Y. Yeh, C. M. Kao, W. P. Sung and C. C. Lin, Curr. Nanosci., 2012, 8, 55–59 CrossRef CAS.
- Y. C. Chang, S. C. Huang and K. F. Chen, Water Sci. Technol., 2014, 69, 2357–2363 CrossRef CAS PubMed.
- I. Hussain, Y. Zhang, S. Huang and X. Du, Chem. Eng. J., 2012, 203, 269–276 CrossRef CAS.
- G.-D. Fang, D. D. Dionysiou, D.-M. Zhou, Y. Wang, X.-D. Zhu, J.-X. Fan, L. Cang and Y.-J. Wang, Chemosphere, 2013, 90, 1573–1580 CrossRef CAS PubMed.
- R. H. Waldemer, P. G. Tratnyek, R. L. Johnson and J. T. Nurmi, Environ. Sci. Technol., 2007, 41, 1010–1015 CrossRef CAS PubMed.
- C. Liang, C. F. Huang and Y. J. Chen, Water Res., 2008, 42, 4091–4100 CrossRef CAS PubMed.
- M. Ahmad, A. L. Teel and R. J. Watts, J. Contam. Hydrol., 2010, 115, 34–45 CrossRef CAS PubMed.
- A. L. Teel, M. Ahmad and R. J. Watts, J. Hazard. Mater., 2011, 196, 153–159 CrossRef CAS PubMed.
- N. Yan, F. Liu, Q. Xue, M. L. Brusseau, Y. Liu and J. Wang, Chem. Eng. J., 2015, 274, 61–68 CrossRef CAS PubMed.
- S. H. Do, J. H. Jo, Y. H. Jo, H. K. Lee and S. H. Kong, Chemosphere, 2009, 77, 1127–1131 CrossRef CAS PubMed.
- S. D. Richardson, B. L. Lebrom, C. T. Miller and M. D. Aitken, Environ. Sci. Technol., 2011, 45, 719–725 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |