
Asperones A–E, five dimeric polyketides with new carbon skeletons from the fungus Aspergillus sp. AWG 1–15†
Xiao-Bing
Wang,  a
Ling-Yi
Kong
*a
and
a
Ling-Yi
Kong
*a
and
a
Ling-Yi
Kong
*a
and
Abstract
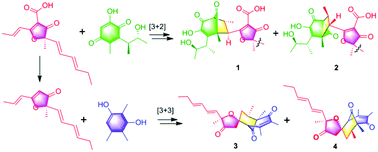
Asperones A–E (1–5), five dimeric polyketides with two distinct skeletons generated by the crucial [3 + 2] and [3 + 3] cycloadditions, were isolated from fungal metabolites of an Aspergillus sp. Their structures were established by spectroscopic data, chemical derivatization, single-crystal X-ray diffraction, and electronic circular dichroism calculations. Compounds 2–4 showed significant nitric oxide (NO) inhibition in lipopolysaccharide (LPS)-induced RAW 264.7 macrophage cells, and exhibited IC50 values of 16.0, 13.2, and 6.0 μM, respectively.
- This article is part of the themed collection: Organic Chemistry Frontiers HOT articles for 2018
 Please wait while we load your content...
Please wait while we load your content...

