
Application of a rhodium-catalyzed cyclization cycloaddition cascade strategy to the total synthesis of (−)-curcumol†
P.
Chiu  *b
*b
*b
Abstract
A de novo asymmetric total synthesis of (−)-curcumol has been realized, based on a rhodium-catalyzed cyclization–cycloaddition cascade reaction as the key step to construct the tricyclic nucleus.
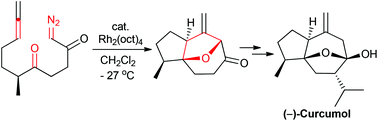
- This article is part of the themed collections: Celebrating Excellence in Research: Women at the Frontiers of Chemistry and Synthetic Approaches to Natural Products via Catalytic Processes
 Please wait while we load your content...
Please wait while we load your content...

