
Isolation, structural elucidation, and synthetic study of salviyunnanone A, an abietane derived diterpenoid with a 7/5/6/3 ring system from Salvia yunnanensis†
Gang
Xu  *a
*a
*a
Abstract
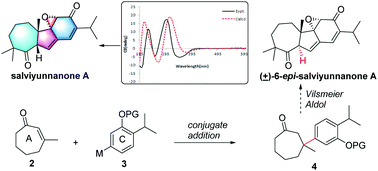
Salviyunnanone A (1), a cytotoxic diterpenoid possessing an unprecedented 7/5/6/3 ring system derived from a normal abietane skeleton, was characterized from Salvia yunnanensis. Its structure was established by extensive MS and NMR spectroscopic analyses and its absolute configuration was defined by the comparison of experimental and calculated ECDs. The synthetic study of 1 was carried out and its 6-epi-isomer (12) was achieved in 9 steps involving the conjugated addition of an aryl boronic acid as a key step.
- This article is part of the themed collection: Organic Chemistry Frontiers HOT articles for 2018
 Please wait while we load your content...
Please wait while we load your content...

