
Racemic trinorsesquiterpenoids from the Beihai sponge Spongia officinalis: structure and biomimetic total synthesis†
 *a
and
Yue-Wei
Guo
*ad
*a
and
Yue-Wei
Guo
*ad
Abstract
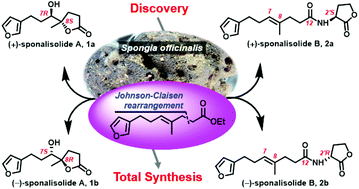
Two rare new furan butanolides, namely sponalisolides A (1) and B (2), characterized by an unprecedented furan-bearing trinorsesquiterpene alkyl chain connecting either a butanolide (1) or an N-acyl homoserine lactone moiety (2), were isolated in racemic forms from the Beihai sponge Spongia officinalis, and were further separated, by chiral-phase HPLC, to their corresponding enantiomers 1a/1b and 2a/2b, respectively. The structures, including the absolute stereochemistry, of the two pairs of enantiomeric compounds, were unambiguously established by extensive spectroscopic analysis and biomimetic total synthesis, involving a key Johnson-Claisen rearrangement and a lactone cyclization after epoxidation or dihydroxylation. All the new compounds exhibited the Pseudomonas aeruginosa quorum sensing inhibitory activity.
- This article is part of the themed collection: Organic Chemistry Frontiers HOT articles for 2018
 Please wait while we load your content...
Please wait while we load your content...

