
ortho-Amino group functionalized 2,2′-bipyridine based Ru(ii) complex catalysed alkylation of secondary alcohols, nitriles and amines using alcohols†
 *a
*a
Abstract
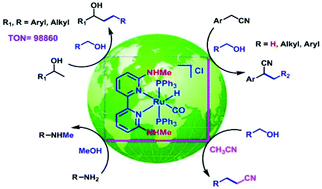
Various Ru(II) complexes bearing functionalized 2,2′-bipyridine ligands were synthesized and fully characterized. Among them, a new N6,N6′-dimethyl-2,2′-bipyridine-6,6′-diamine ligand was found to be the most electron-rich ligand as its corresponding Ru(II) complex (1a) displayed the lowest νco value and the highest efficiency in the β-alkylation of secondary alcohols with primary alcohols (TON = 98 860). Complex 1a also exhibited a greater reactivity in the monoalkylation of acetonitrile, α-alkylation as well as α-methylation of arylacetonitriles. Compared to the other reported systems, in α-methylation of nitriles complex 1a presented superior catalytic activity. The potential of complex 1a was extended further in N-methylation of amines using methanol as a green methylating agent.
- This article is part of the themed collection: Organic Chemistry Frontiers HOT articles for 2018
 Please wait while we load your content...
Please wait while we load your content...

