
Synthesis of pyridine trans-tetrafluoro-λ6-sulfane derivatives via radical addition†
Etsuko
Tokunaga,  a
Norio
Shibata
*a
a
Norio
Shibata
*a
a
Norio
Shibata
*a
Abstract
Pyridine tetrafluoro-λ6-sulfanes (SF4) with alkenyl or alkyl substituents have been synthesized for the first time via the radical addition reactions of pyridine-SF4 chlorides to alkynes or alkenes in good to high yields. X-Ray crystallographic analysis and DFT calculations disclosed an octahedral symmetrical trans-configuration of the hypervalent tetrafluorosulfanyl center. In contrast to phenyl-SF4 analogues, pyridine-SF4 compounds were found to be stable, which expands the utility of pyridine-SF4 compounds.
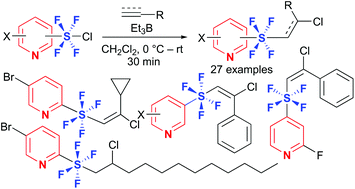
- This article is part of the themed collections: Organic Chemistry Frontiers HOT articles for 2018 and Organic Chemistry Frontiers HOT articles for 2017
 Please wait while we load your content...
Please wait while we load your content...

