
Regioselective C5 alkenylation of 2-acylpyrroles via Pd(II)-catalyzed C–H bond activation†
Jian-Hua
Duan
,
Rui-Jie
Mi
,
Jing
Sun
* and
Ming-Dong
Zhou
 *
*
School of Chemistry and Materials Science, Liaoning Shihua University, Dandong Road 1, Fushun 113001, P. R. China. E-mail: sunjing@lnpu.edu.cn; mingdong.zhou@lnpu.edu.cn
First published on 9th October 2017
Abstract
A Pd(II)-catalyzed regioselective C5 alkenylation of 2-acylpyrroles has been developed employing 3-nitrile benzoyl group (N-(3-NCC6H4CO-)) as an efficient N-protecting group. The protocol provided a simple and efficient method to synthesize C5-alkenylated 2-acylpyrrole derivatives. The employed N-protecting group was readily removable in the presence of HCl/EtOH under mild conditions.
Introduction
Pyrrole is one of the most important heterocyclic compounds and is the ubiquitous backbone in natural products, bioactive molecules, pharmaceutical compounds and organic materials.1–4 In particular, 2-acylpyrrole-containing heterocyclic compounds have attracted much attention in medical chemistry5–8 because the 2-acylpyrrole moiety is a key unit found in many pharmaceutical compounds.9,10 For example, Tolmetin and Zomepirac are non-steroidal anti-inflammatory drugs, and the other 2-acylpyrrole derivatives also exhibit excellent anti-inflammatory activity as potential drugs (Fig. 1).8,11 Synthetic chemists have confirmed that 2-acylpyrroles possess interesting biological and biomedical properties.12 Moreover, 2-arylpyrroles are also of interest in the synthesis of fluorescent dyes and insecticides.13 The most common route to pyrrole derivatives involves cyclization reactions, such as the Knorr, Paal–Knorr, and Hantzsch reactions.14 Unfortunately, these methods often require robust and versatile transformations. In this regard, the development of an efficient strategy for the functionalization of 2-acylpyrrole is attractive and desirable.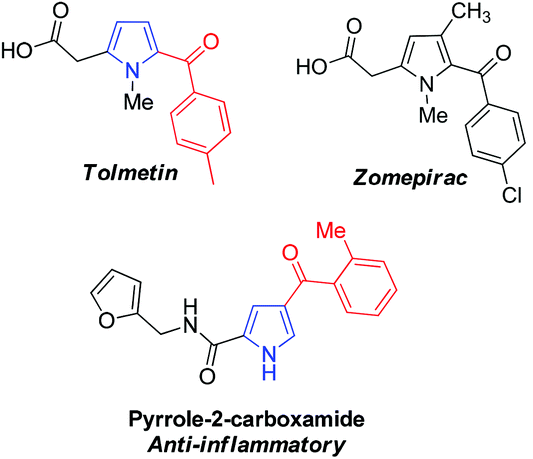 | ||
| Fig. 1 2-Acylpyrrole in pharmaceuticals. | ||
Recent progress in transition metal catalyzed direct C–H bond activation has opened up high atom- and step-economy routes to substituted heterocyclic compounds.15–17 Amongst various transformations, the C–H functionalization of indole derivatives at different positions employing palladium, ruthenium or rhodium complexes as catalysts has attracted much attention.18 In contrast, the C–H bond activation of pyrrole derivatives has been less explored, probably due to its instability under acidic and oxidizing environments that limit its utility in metal catalyzed C–H transformations.19,20 Despite this fact, some elegant methods including C–H arylation, alkenylation, and borylation have been developed, which primarily focus on C2 and/or C5 functionalization of pyrrole. However, very few studies have been reported for functionalization at the C3-position.19,20 For example, Gaunt et al.19a have developed a mild aerobic oxidative palladium(II) catalyzed pyrrole C–H bond functionalization by adjusting the N protecting groups with different steric and electronic properties, in which either C2 or C3 alkenylation products were obtained in good yield. Yao et al.20d recently reported a solvent-controlled switchable C–H alkenylation of 3,4-disubstituted pyrrole via palladium(II) catalyzed oxidation. Heck reaction, enabling C2 or C5 alkenylation products, was also applied to the total synthesis of the natural product (±)-rhazinilam. Encouraged by these protocols, we speculated that the palladium-catalyzed direct C5 alkenylation of 2-acylpyrrole derivatives should be feasible with the assistance of a suitable N-protecting group. To the best of our knowledge, the direct C5 alkenylation of 2-acylpyrroles via palladium-catalyzed C–H bond activation has not been reported yet. Herein, we wish to disclose our development on the palladium-catalyzed regioselective C5 functionalization of 2-acylpyrroles to synthesize C5 alkenylated 2-acylpyrrole derivatives. The products obtained in our study could serve as precursors to prepare the synthetic histone deacetylase (HDAC) inhibitors.21
Initially, phenyl(1H-pyrrol-2-yl)methanone and ethyl acrylate were applied as model substrates to explore their reactivity. As predicted, when using the unprotected 2-acylpyrrole-phenyl(1H-pyrrol-2-yl)methanone, the reaction afforded complicated products and no desired alkenylated product could be isolated under the condition of 20 mol% Pd(OAc)2 and 3 equiv. AgOAc in toluene at 130 °C for 12 h (entry 1, Table 1). The first attempt using tosyl (N-Ts) as the protecting group was not successful. To our delight, further studies using N-(3-NCC6H4CO-) protected 2-acylpyrrole afforded 30% of C5-alkenylated products (entries 2 and 3, Table 1). Further, the reaction using 3-(2-benzoyl-1H-pyrrole-1-carbonyl)benzonitrile (1a) was optimized. Screening of different solvents indicated that the solvent had a significant impact on the C–H activation, and PhCF3 turned out to be the best solvent (entries 4–9, Table 1). Furthermore, varying the reaction temperature, also varied the yield, and a good yield (70%) could be achieved on lowering the reaction temperature to 100 °C (entries 10–12, Table 1). On reducing the amount of AgOAc from 3 equiv. to 2 equiv., the yield reduced to 60% (entry 13, Table 1). However, increasing the amount of AgOAc to 4 equiv. could not further improve the product yield (entry 14, Table 1). Moreover, 20 mol% Pd(OAc)2 proved to be necessary in order to achieve a good activity for the C–H functionalization, which could be due to the deactivation effect that arises from the strong electron-withdrawing 2-acyl group (entries 10 and 15–16, Table 1). Accordingly, an optimal reaction condition (Pd(OAc)2 (20 mol%), AgOAc (3 equiv.), PhCF3 (2 mL), 100 °C, 12 h) was obtained. Based on this, the alkenylation using different protecting groups including 3-NCC6H4CH2-, Boc, and Ts were examined under the optimal condition, affording 60%, 65% (C5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C4 = 1.2:1) and 44% of the desired alkenylation products, respectively (entries 17–19, Table 1). Clearly, the protecting group affects both the reactivity and selectivity; thus, 3-NCC6H4CO- proved to be favorable for this alkenylation reaction.
:C4 = 1.2:1) and 44% of the desired alkenylation products, respectively (entries 17–19, Table 1). Clearly, the protecting group affects both the reactivity and selectivity; thus, 3-NCC6H4CO- proved to be favorable for this alkenylation reaction.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R | Solvent | AgOAc (equiv.) | Pd(OAc)2 (mol%) | T (°C) | Yieldb (%) |
|
a Reaction conditions: 3-(2-Benzoyl-1H-pyrrole-1-carbonyl)benzonitrile 1a (0.2 mmol), ethyl acrylate (0.3 mmol), Pd(OAc)2, 2 mL of solvent, 12 h.
b Isolated yield.
c The reaction afforded a mixture of C4 and C5-alkenylated products (C5:C4 = 1.2:1).
|
||||||
| 1 | H | Toluene | 3 | 20 | 130 | 0 |
| 2 | Ts | Toluene | 3 | 20 | 130 | 0 |
| 3 | 3-NCC6H4CO- | Toluene | 3 | 20 | 130 | 30 |
| 4 | 3-NCC6H4CO- | DCE | 3 | 20 | 130 | 0 |
| 5 | 3-NCC6H4CO- | TFA | 3 | 20 | 130 | 0 |
| 6 | 3-NCC6H4CO- | DMF | 3 | 20 | 130 | 0 |
| 7 | 3-NCC6H4CO- | Benzene | 3 | 20 | 130 | 30 |
| 8 | 3-NCC6H4CO- | C6F6 | 3 | 20 | 130 | 48 |
| 9 | 3-NCC6H4CO- | PhCF3 | 3 | 20 | 130 | 50 |
| 10 | 3-NCC6H4CO- | PhCF3 | 3 | 20 | 100 | 70 |
| 11 | 3-NCC6H4CO- | PhCF3 | 3 | 20 | 80 | 38 |
| 12 | 3-NCC6H4CO- | PhCF3 | 3 | 20 | 40 | 18 |
| 13 | 3-NCC6H4CO- | PhCF3 | 2 | 20 | 100 | 60 |
| 14 | 3-NCC6H4CO- | PhCF3 | 4 | 20 | 100 | 65 |
| 15 | 3-NCC6H4CO- | PhCF3 | 3 | 10 | 100 | 46 |
| 16 | 3-NCC6H4CO- | PhCF3 | 3 | 5 | 100 | 41 |
| 17 | 3-NCC6H4CH2- | PhCF3 | 3 | 20 | 100 | 60 |
| 18 | Ts | PhCF3 | 3 | 20 | 100 | 65c |
| 19 | Boc | PhCF3 | 3 | 20 | 100 | 44 |
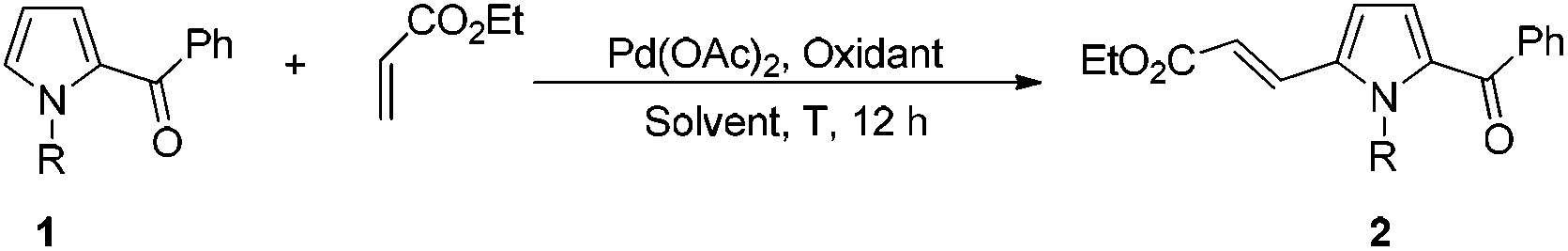
With the optimized reaction conditions in hand, the scope and generality of this transformation was investigated (Tables 2 and 3). The results indicated that the aromatic units with different substituents were amenable for this transformation (Table 2). For the 2-acylpyrroles bearing electron-donating methyl group on the acyl phenyl ring, the reaction afforded the desired products in moderate yield. The influence of steric effect on the reaction activity was not observed (2b–2d). Comparatively lower yields were obtained with methoxy substituted 2-acylpyrroles (2e, 2f). Substrates with halogen group (F, Cl, Br) at ortho, meta, or para positions of acyl phenyl ring all reacted smoothly to afford the corresponding products in 50–68% yields (2g–2m). Furthermore, 2-acylpyrroles bearing the strong electron withdrawing CF3 or CO2Me group on the acyl phenyl ring also provided satisfactory yields of the C5-alkenylated products (2n–2o). We attempted to examine the reactivity of substrates with disubstituents on the acyl phenyl moiety, which were also smoothly transformed to the desired products in moderate yields (2p–2s).
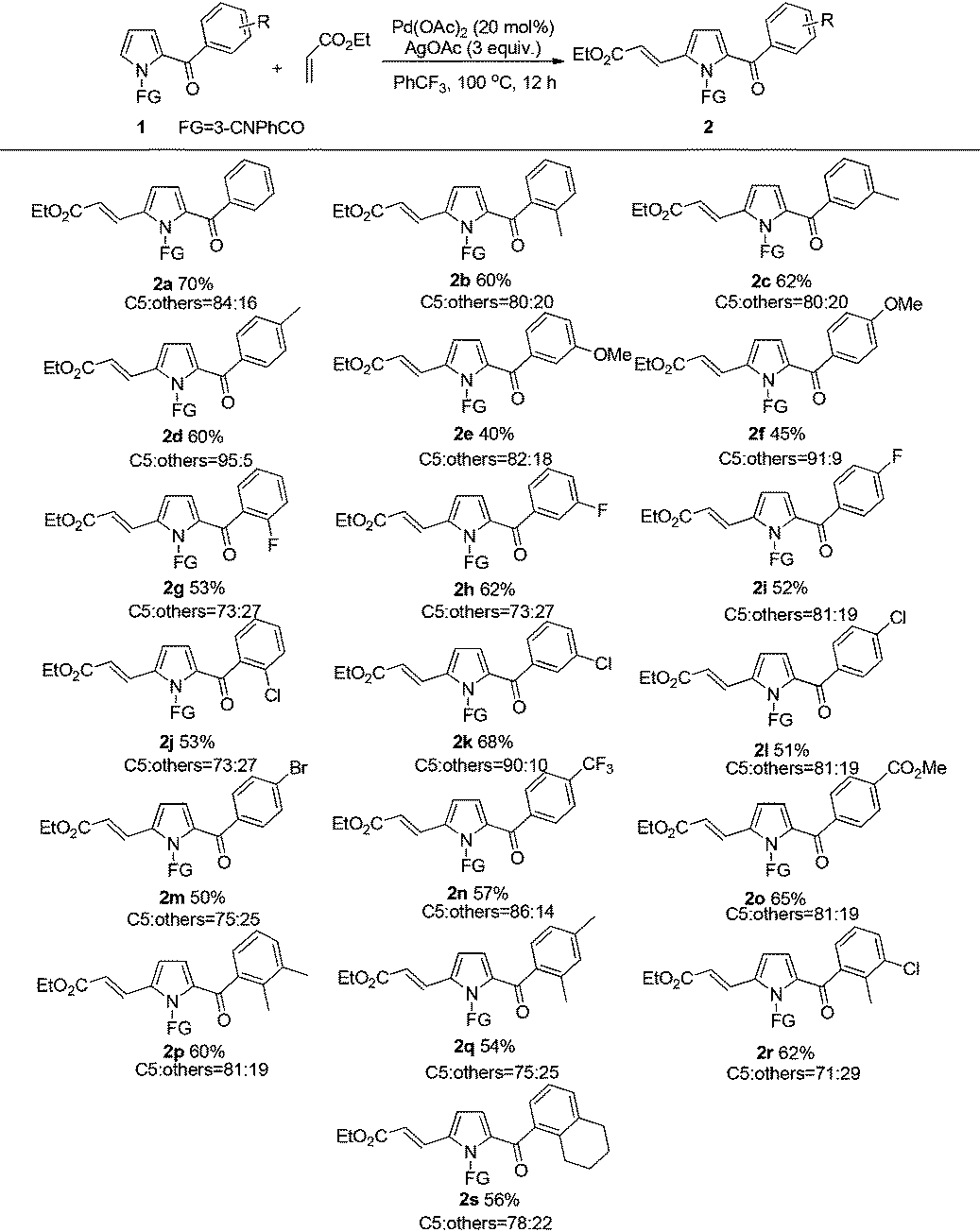
Next, the scope for alkene as a substrate was examined (Table 3). It was satisfactory to observe that the scope of this reaction could also be extended to other alkenes such as methyl acrylate, 2,2,2-trifluoroethyl acrylate, (phenylsulfonyl)ethene, acrylonitrile and acrolein, albeit in somewhat lower product yields (3a–3e). Nevertheless, the reaction using diethyl vinylphosphonate and 4-(trifluoromethyl)styrene as the coupling partner only led to complicated and inseparable product mixtures. Moreover, the alkenylation using 1-acetylcyclohexene proved to be unreactive, which could be due to the strong steric effect of cyclohexene ring that hindered the addition of alkene to the Pd–C bond of the palladation intermediate for this C–H alkenylation.
To further demonstrate the potential of the developed method, a gram-scale synthesis of compound 2a under the standard conditions was conducted. Reaction of 1a (5 mmol) with ethyl acrylate afforded the target product 2a in 60% yield (Scheme 1). As a practical industrial application, it is also important to remove the protecting group. To our delight, the N-pyrrole protecting group could be easily removed on treating 2a with HCl/EtOH (1:5) for 3 h at 90 °C, yielding 68% of the (NH)-free product 4 (Scheme 2).
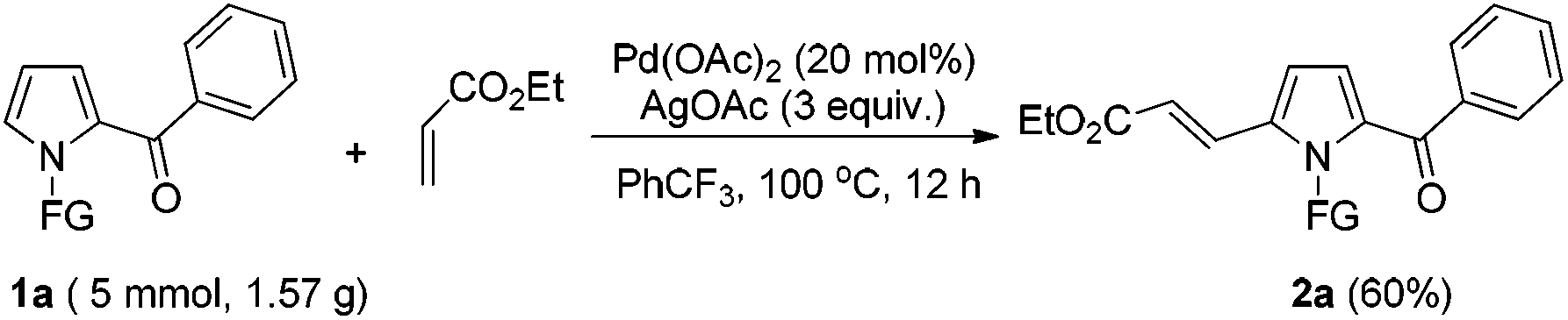 | ||
| Scheme 1 Gram-scale synthesis of 2a. | ||
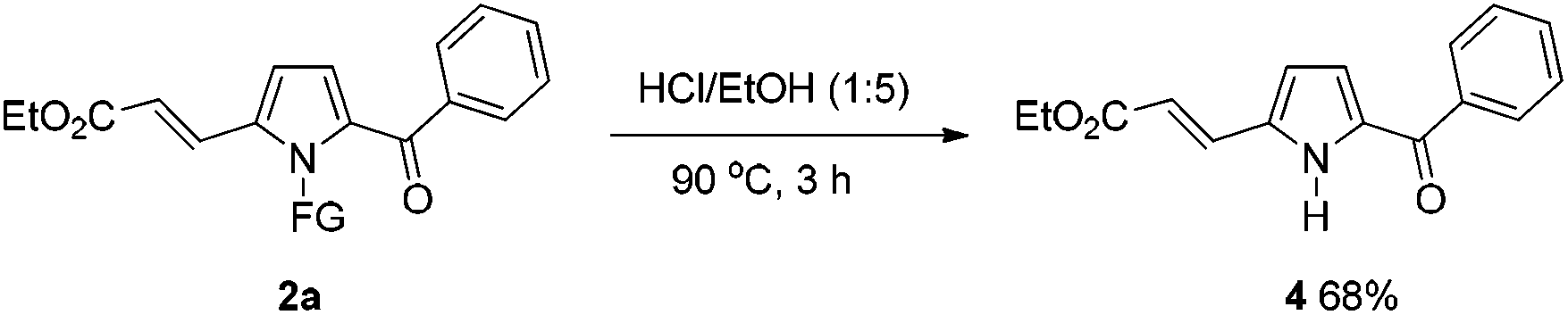 | ||
| Scheme 2 The removal of N protecting group. | ||
Based on the experimental results and literature precedents,19a,20d a possible mechanism was proposed as shown in Scheme 3. Initially, the 2-acylpyrrole substrate 1 reacts with palladium catalyst through an electrophilic C–H activation pathway at the C5 position of pyrrole to afford an intermediate A. Then, the dearomatization of A results in the formation of the palladation intermediate B. The subsequent addition of alkenyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond to Pd–C bond of B, and then the β-H elimination of C leads to a final C5-alkenylated product 2, which undergoes the classical Heck-type reaction. Moreover, the directing effect of carbonyl group of N-3-NCC6H4CO- should not be ignored. As observed, the reaction using 3-NCC6H4CH2- as protecting group demonstrated a lower yield of 2a (60%) than that using tosyl (N-Ts) (65%) and N-3-NCC6H4CO- (70%) (Table 1). However, such directing (or coordinating) effect of protecting groups should not be dominant for this C5-alkenylation.
C double bond to Pd–C bond of B, and then the β-H elimination of C leads to a final C5-alkenylated product 2, which undergoes the classical Heck-type reaction. Moreover, the directing effect of carbonyl group of N-3-NCC6H4CO- should not be ignored. As observed, the reaction using 3-NCC6H4CH2- as protecting group demonstrated a lower yield of 2a (60%) than that using tosyl (N-Ts) (65%) and N-3-NCC6H4CO- (70%) (Table 1). However, such directing (or coordinating) effect of protecting groups should not be dominant for this C5-alkenylation.
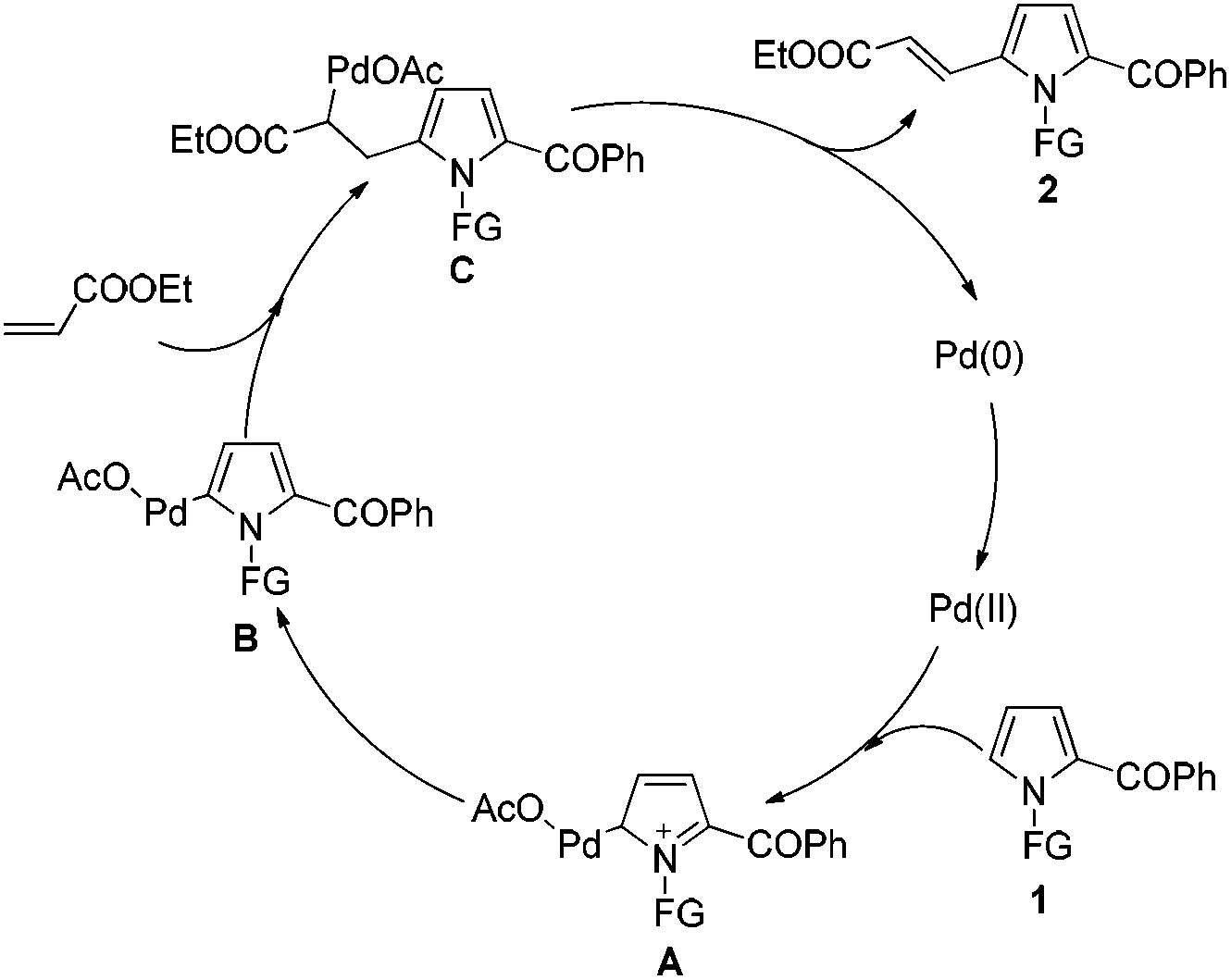 | ||
| Scheme 3 Proposed mechanism for C5 alkenylation of 2-acylpyrroles. | ||
Conclusions
In summary, we have developed a palladium(II)-catalyzed direct C5 alkenylation of 2-acylpyrroles using various functionalized alkenes as coupling partners. This transformation shows good C5 selectivity and is tolerant towards various acyl phenyl substituted 2-acylpyrroles, affording a wide range of 2,5-disubstituted pyrrole derivatives as the only isolated products. The products could serve as potential pharmaceuticals, in particular, as precursors to prepare the synthetic histone deacetylase (HDAC) inhibitors. In addition, the N-pyrrole protecting group is easily removed to obtain the (NH)-free pyrrole compounds under mild conditions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Science Foundation of China (21101085), Natural Science Foundation of Liaoning Province (2015020196), Research Fund of Key Laboratory of Synthetic Rubber – Changchun Institute of Applied Chemistry (KLSR1602), Talent Scientific Research Fund of LSHU (2016XJJ-006), and Research Project Fund of Liaoning Provincial Department of Education (L2016022) for the financial supports.Notes and references
- M. Stepien, E. Gonka, M. Zyla and N. Sprutta, Chem. Rev., 2017, 117, 3479 CrossRef CAS PubMed.
- A. Gulevich, A. Dudnik, N. Chernyak and V. Gevorgyan, Chem. Rev., 2013, 113, 3084 CrossRef CAS PubMed.
- (a) C. B. Bheeter, L. Chen, J. F. Soule and H. Douct, Catal. Sci. Technol., 2016, 6, 2005 RSC; (b) M. Kangani, N. Hazeri and M. T. Maghsoodlou, J. Saudi Chem. Soc., 2017, 21, 160 CrossRef CAS.
- (a) R. Tanaka, H. Ikemoto, M. Kanai, T. Yoshino and S. Masunafa, Org. Lett., 2016, 18, 5732 CrossRef CAS PubMed; (b) S. Maddila, S. Gorle, Ch. Sampath and P. Lavanya, J. Saudi Chem. Soc., 2016, 20, s306 CrossRef CAS; (c) R. Rossi, F. Bellina, M. Lessi and C. Manzini, Adv. Synth. Catal., 2014, 356, 17 CrossRef CAS.
- S. Etcheverry, D. Barrio, A. Cortizo and P. Willams, J. Inorg. Biochem., 2002, 88, 94 CrossRef CAS PubMed.
- M. Yuan, M. Luo, Y. Song, Q. Xu, X. Wang, Y. Cao, X. Bu, Y. Ren and X. Hu, Bioorg. Med. Chem., 2011, 19, 1189 CrossRef CAS PubMed.
- Z. Guo, X. Wei, Y. Hua, J. Chao and D. Liu, Tetrahedron Lett., 2015, 56, 3919 CrossRef CAS.
- R. Khajuria, S. Dham and K. Kapoor, RSC Adv., 2016, 6, 37039 RSC.
- (a) S. Tamaru, L. Yu, W. Youngblood, K. Muthukumaran, M. Taniguchi and J. Lindsey, J. Org. Chem., 2004, 69, 765 CrossRef CAS PubMed; (b) S. Zaidi, K. Muthukumaran, S. Tamaru and J. Lindsey, J. Org. Chem., 2004, 69, 8356 CrossRef CAS PubMed; (c) R. Rieth, N. Mankad, E. Calimano and J. Sadighi, Org. Lett., 2004, 6, 3981 CrossRef CAS PubMed.
- (a) R. Wakeham, J. Taylor, S. Bull, J. Morris and J. Williams, Org. Lett., 2013, 15, 702 CrossRef CAS PubMed; (b) H. Kikuchi, M. Sekiya, Y. Katou, K. Ueda, T. Kabeya, S. Kurata and Y. Oshima, Org. Lett., 2009, 11, 1693 CrossRef CAS PubMed.
- F. Jafarpour, H. Hazrati and M. Darvishmolla, Adv. Synth. Catal., 2014, 356, 3784 CrossRef CAS.
- (a) Y. Liu, N. Haste, W. Thienphrapa, V. Nizet, M. Hensler and R. Li, Mar. Drugs, 2012, 10, 953 CrossRef CAS PubMed; (b) K. Yamanaka, K. Ryan, T. Gulder, C. Hughes and B. Moore, J. Am. Chem. Soc., 2012, 134, 12434 CrossRef CAS PubMed.
- (a) A. Burghart, L. H. Thoresen, J. Chen, K. Burgess, F. Bergström and L. B.-Å. Johansson, Chem. Commun., 2000, 2203 RSC; (b) G. A. Pinna, G. Loriga, G. Murineddu, G. Grella, M. Mura, L. Vargiu, C. Murgioni and P. La Colla, Chem. Pharm. Bull., 2001, 49, 1406 CrossRef CAS PubMed; (c) D. A. Hunt, Pestic. Sci., 1996, 47, 201 CAS.
- (a) A. Wanger and M. Sanford, Org. Lett., 2011, 13, 288 CrossRef PubMed; (b) C. Schmuck and D. Rupprecht, Synthesis, 2007, 3095 CrossRef CAS.
- (a) L. Ping, D. Chung, J. Bouffard and S. Lee, Chem. Soc. Rev., 2017, 46, 4299 RSC; (b) R. Y. Zhu, M. E. Farmer, Y. Q. Chen and J.-Q. Yu, Angew. Chem., Int. Ed., 2016, 55, 10578 CrossRef CAS PubMed; (c) H. Huang, X. Ji, W. Wu and H. Jiang, Chem. Soc. Rev., 2015, 44, 1155 RSC; (d) G. Qiu and J. Wu, Org. Chem. Front., 2015, 2, 169 RSC.
- (a) Y. Liang and N. Jiao, Acc. Chem. Res., 2017, 50, 1640 CrossRef CAS PubMed; (b) K. Shin, H. Kim and S. Chang, Acc. Chem. Res., 2015, 48, 1040 CrossRef CAS PubMed; (c) B. H. Ye and N. Cramer, Acc. Chem. Res., 2015, 48, 1308 CrossRef CAS PubMed; (d) K. M. Engle, T. S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed.
- (a) H.-W. Wang, Y. Lu, B. Zhang, J. He, H.-J. Xu, Y.-S. Kang, W.-Y. Sun and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 7449 CrossRef CAS PubMed; (b) H.-J. Xu, Y. Lu, M. E. Farmer, H.-W. Wang, D. Zhao, Y.-S. Kang, W.-Y. Sun and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 2200 CrossRef CAS PubMed; (c) Y. Lu, H.-W. Wang, J. E. Spangler, K. Chen, P.-P. Cui, Y. Zhao, K. W.-Y. Sun and J.-Q. Yu, Chem. Sci., 2015, 6, 1923 RSC; (d) K. Ueda, K. Amaike, R. Maceiczyk, K. Itami and J. Yamaguchi, J. Am. Chem. Soc., 2014, 136, 13226 CrossRef CAS PubMed; (e) B. Li, J. Ma, W. Xie, H. Song, S. Xu and B. Wang, Chem. – Eur. J., 2013, 19, 11863 CrossRef CAS PubMed; (f) L. Jiao and T. Bach, Angew. Chem., Int. Ed., 2013, 52, 6080 CrossRef CAS PubMed; (g) B. Li, J. Ma, W. Xie, H. Song, S. Xu and B. Wang, J. Org. Chem., 2013, 78, 9345 CrossRef CAS PubMed; (h) Y. Lu, D.-H. Wang, K. M. Engle and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 5916 CrossRef CAS PubMed; (i) S. H. Cho, S. J. Hwang and S. Chang, J. Am. Chem. Soc., 2008, 130, 9254 CrossRef CAS PubMed; (j) M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 14058 CrossRef CAS PubMed; (k) D. H. Wang, T. S. Mei and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 17676 CrossRef CAS PubMed; (l) J. J. Li, T. S. Mei and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 6452 CrossRef CAS PubMed; (m) X. Wang, D. Gribkov and D. Sames, J. Org. Chem., 2007, 72, 1476 CrossRef CAS PubMed.
- (a) C. Della, M. Fontana, E. Motti and M. Catellani, Acc. Chem. Res., 2016, 49, 1389 CrossRef PubMed; (b) A. Sandorv, Adv. Synth. Catal., 2015, 357, 2403 CrossRef; (c) L. Ackermann, Acc. Chem. Res., 2014, 47, 281 CrossRef CAS PubMed; (d) L. Xu, C. Zhang, Y. He, L. Tan and D. Ma, Angew. Chem., Int. Ed., 2016, 55, 321 CrossRef CAS PubMed; (e) Y. Yang, X. Qiu, Y. Zhao, Y. Mu and Z. Shi, J. Am. Chem. Soc., 2016, 138, 495 CrossRef CAS PubMed; (f) Y. Yang, R. Li, Y. Zhao, D. Zhao and Z. Shi, J. Am. Chem. Soc., 2016, 138, 8734 CrossRef CAS PubMed; (g) B. S. Lane, M. A. Brown and D. Sames, J. Am. Chem. Soc., 2005, 127, 8050 CrossRef CAS PubMed; (h) N. P. Grimster, C. D. Godfrey and M. Gaunt, Angew. Chem., Int. Ed., 2005, 44, 3125 CrossRef CAS PubMed; (i) E. M. Ferreira and B. M. Stoltz, J. Am. Chem. Soc., 2003, 125, 9578 CrossRef CAS PubMed.
- (a) E. Beck, N. Grimster, R. Hatley and M. Gaunt, J. Am. Chem. Soc., 2006, 128, 2528 CrossRef CAS PubMed; (b) A. García-Rubia, R. Arrayás and J. Carretero, Angew. Chem., Int. Ed., 2009, 48, 6511 CrossRef PubMed.
- (a) A. Garcia-Rubia, B. Urones, R. Arrayas and J. Carretero, Chem. – Eur. J., 2010, 16, 9676 CrossRef CAS PubMed; (b) J. Brand, J. Charpentier and J. Waser, Angew. Chem., Int. Ed., 2009, 48, 9346 CrossRef CAS PubMed; (c) I. Ban, T. Sudo, T. Taniguchi and K. Itami, Org. Lett., 2008, 10, 3607 CrossRef CAS PubMed; (d) Y. Su, H. Zhou, J. Chen, J. Xu, X. Wu, A. Lin and H. Yao, Org. Lett., 2014, 16, 4884 CrossRef CAS PubMed.
- R. Ragno, A. Mai, S. Massa, I. Cerbara, S. Valente, P. Bottoni, R. Scatena, F. Jesacher, P. Loidl and G. Brosch, J. Med. Chem., 2004, 47, 1351 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectral data for all compounds. See DOI: 10.1039/c7qo00732a |
| This journal is © the Partner Organisations 2018 |