
Scandium-catalyzed C(sp3)–H alkylation of N,N-dimethyl anilines with alkenes†
Hongjie
Gao
,
Jianhong
Su
,
Pengfei
Xu
and
Xin
Xu
 *
*
Key Laboratory of Organic Synthesis of Jiangsu Province, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, P. R. China. E-mail: xinxu@suda.edu.cn
First published on 20th September 2017
Abstract
A scandium complex based on a new type of tridentate ligand enabled an atom- and step-economical C(sp3)–H addition of N,N-dimethyl anilines to a variety of unactivated alkenes affording branched products for the first time. A cationic o-dimethylaminophenyl scandium species was isolated and confirmed as the catalytic intermediate in this transformation.
Aromatic amines are ubiquitous in chemical science and play important roles in the pharmaceutical and fine chemical industries.1 Great efforts have been made to develop selective and efficient approaches for the synthesis of aromatic amines. To this end, catalytic hydroamination2 and reductive amination3 were explored and successfully utilized. In both reactions, the presence of an N–H bond in the starting amine is essential for the formation of a new C–N bond. Recently, direct functionalization of the inert C–H bond has flourished4 and has provided an alternative and ideal way for the preparation of aromatic amines from simple anilines. Compared with cross dehydrogenative coupling via oxidation processes5 or carbene insertion6 in the area, the transition-metal catalyzed C–H addition of aniline derivatives to readily available alkenes has attracted increased interest as it offers a 100% atom economical method to various alkylated anilines.7 However, late transition-metal catalyzed Friedel–Crafts type reactions of anilines with activated alkenes, e.g. styrene, usually gave a mixture of ortho- and para-alkylation products.8,9 Recently, Hou and co-workers reported a highly efficient ortho-selective C(sp2)–H alkylation of tertiary aniline with alkenes promoted by a half-sandwich yttrium catalyst (Scheme 1a).10 Furthermore, Hartwig11 and others12 have made significant progress in the α-C(sp3)–H alkylation of N-alkyl anilines with alkenes, namely hydroaminoalkylation, by using group 4 and group 5 catalysts (Scheme 1b). However, for the α-C(sp3)–H alkylation of anilines, so far the substrate scope is still limited in primary and secondary amines. To date, the catalytic C(sp3)–H alkylation of tertiary anilines with alkenes remains scarce.13,14 We herein report a highly efficient and regioselective C(sp3)–H addition of N,N-dimethyl tertiary anilines towards a variety of unactivated alkenes for the first time, which is catalyzed by a scandium complex based on a novel β-diketiminato ligand (Scheme 1c). Mechanistic studies revealed that a cationic o-dimethylaminophenyl scandium complex served as the catalytic intermediate and the conversion of ortho-C(sp2)–H metalation to α-C(sp3)–H metalation may be involved in the catalytic process leading to the formation of unexpected hydroaminoalkylation products.
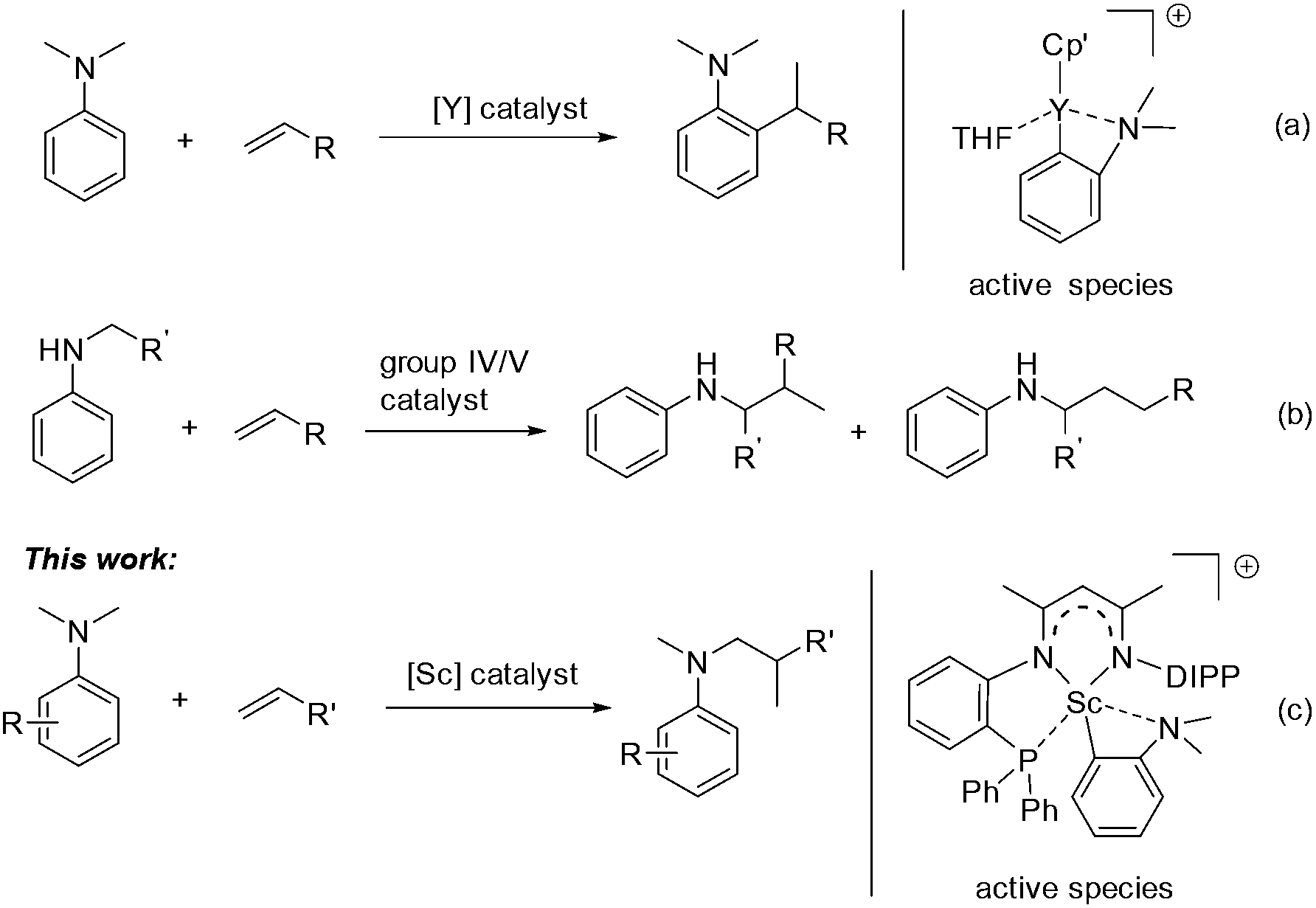 | ||
| Scheme 1 Selected examples of the C–H addition of anilines to alkenes. | ||
We recently designed a new type of β-diketiminato ligand with a pendant phosphine group and prepared the corresponding scandium dialkyl complex 1 as the precursor for the rare-earth metal based frustrated Lewis pair.15 As continued interest in this area, we prepared an analogous β-diketiminato ligand bearing a rigid phosphine arm along with the corresponding rare-earth metal dialkyls 2 (Sc) and 3 (Y).16 We initiated our studies by employing a series of β-diketiminato rare-earth dialkyls17 together with one equivalent of [PhNHMe2][B(C6F5)4] for the reaction of N,N-dimethyl aniline with 1-octene and the results are summarized in Table 1. Catalyst screening clearly showed that the catalytic behavior was highly dependent on the metal ion and the ligand. Only the Sc complex 2 exhibited high activity, exclusively affording the branched C(sp3)–H alkylation product 7aa in 79% isolated yield (8 h, 120 °C, toluene as the solvent, Table 1, entry 2).18 The other three rare-earth complexes showed negligible activity and the Y complex 3 produced trace amounts of the arene C(sp2)–H alkylation product 8aa (Table 1, entry 3), which was the exclusive product in Hou's work catalyzed by a mono-Cp Y complex.10 Therefore, the combination of our scandium complex 2 with [PhNHMe2][B(C6F5)4] showed unique and distinctive catalytic behavior in contrast with the widely investigated cationic half-sandwich rare-earth system.19
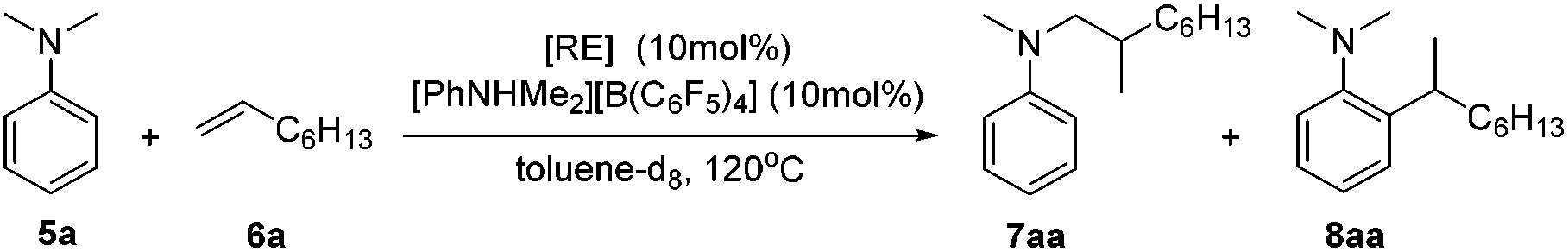

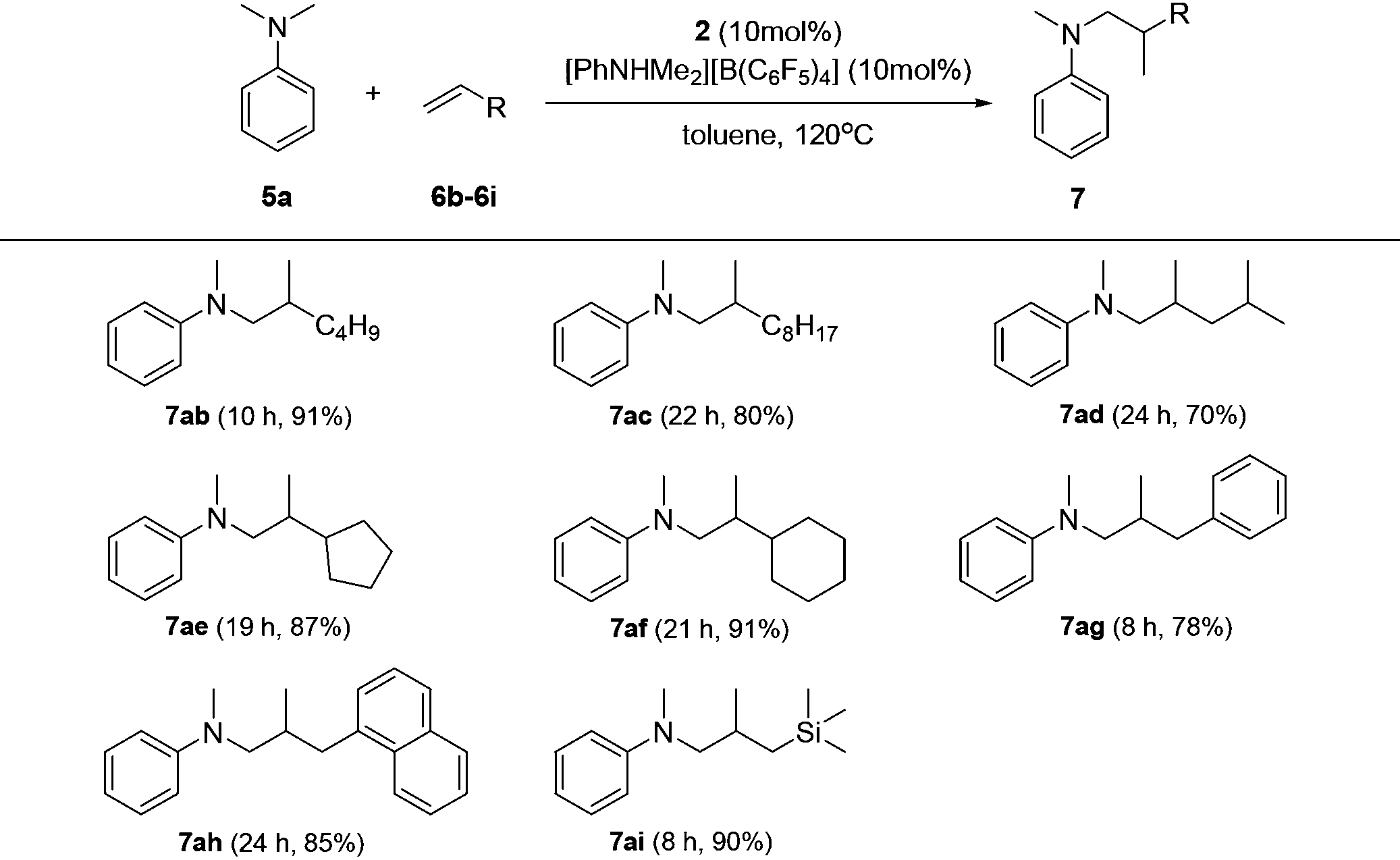
Based on catalyst screening results, we subsequently examined reactions between N,N-dimethyl aniline (5a) with a variety of alkenes promoted by the 2/[PhNHMe2][B(C6F5)4] system at 120 °C in toluene (Table 2). With 10 mol% catalyst loading, simple and unactivated α-olefins, such as 1-hexene (6b), 1-decene (6c) and 4-methyl-1-pentene (6d) were successfully involved in the reactions and gave hydroaminalkylation products 7ab–7ad in good to excellent isolated yields. For more sterically demanding cycloalkyl substituted alkenes 6e and 6f, similar activity and selectivity were observed. Reactions with allylic substrates also took place to exclusively produce the corresponding C(sp3)–H functionalized compounds 7ag–7ai in a short time. When using disubstituted or internal alkenes, e.g. 2-ethyl-1-butene, cis-3-hexene and cyclohexene, there was no detectable alkylation product formed even with a prolonged time (48 h) under the given reaction conditions, which is probably due to the severer steric hindrances in the alkenes. Attempts to use styrene as an alkene substrate led to the polymerization of styrene20 and no expected product was formed. Alkenes containing polar functional groups e.g. acrylonitrile and allyldimethylamine were also not applicable in the reactions.
| a Reaction conditions: 5a (0.71 mmol), 6b–6i (0.47 mmol), toluene (2.5 mL), isolated yield. |
|---|
|
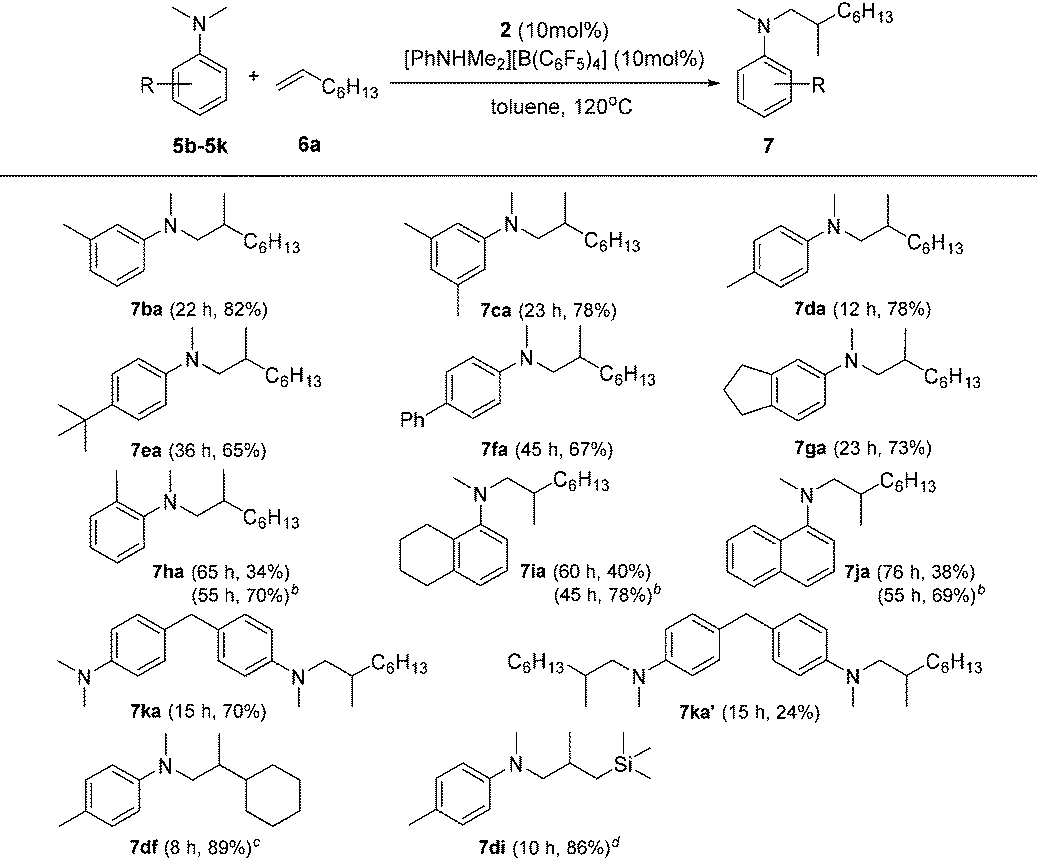
Subsequently, the scope of tertiary aniline was explored under the given reaction conditions (10 mol% catalyst loading, toluene, 120 °C) and the results are shown in Table 3. It is gratifying to notice that a wide range of meta- and para-substituted N,N-dimethyl anilines 5b–5g underwent regioselectively N-methyl C–H alkylation with 1-octene affording corresponding branched addition products in 65–82% isolated yields. The substituents at the ortho-positions on the phenyl ring of the amines are found to slow down (for anilines 5h–5j requiring 20% catalyst loading) or even shut off (for Mes-NMe2; Mes = 2,4,6-trimethylphenyl) the reactions. These findings indicate that the ortho substituent in N,N-dimethyl anilines has a significant influence on this C(sp3)–H alkylation reaction. In the case of 4,4′-methylenebis(N,N-dimethylaniline) (5k), it produced a mixture of the monoalkylation product 7ka (70% yield) and the dialkylation product 7ka′ (24% yield) in 15 h under the standard conditions. Increased yield for the dialkylation product 7ka′ could be achieved by increasing the amount of the starting alkene (Table S4†). It's also noted that the reactions of N,N-dimethyl-p-toluidine (5d) with terminal alkenes 6f and 6i gave N-Me alkylation products in 89% and 86% yields, respectively. Attempts to use methoxyl- or bromide-substituted dimethylaniline as an amine coupling partner failed probably due to the strong oxygen and halogen affinity of the rare-earth metal ion. The C(sp3)–H alkylation reaction of N-methylaniline or N-ethyl-N-methylaniline with 1-octene didn't take place under the standard conditions.
To gain more insight into the active species and reaction mechanism, we investigated the stoichiometric reaction between the Sc dialkyl complex 2 and equimolar [PhNHMe2][B(C6F5)4] in toluene (Scheme 2). After workup, the cationic o-dimethylaminophenyl scandium complex 9 was isolated in 92% yield as expected, which was characterized by multiple nuclear NMR spectroscopy procedures as well as elemental analysis.16 We proposed that the formation of 9 results from the ortho C(sp2)–H activation of N,N-dimethylaniline by a cationic monoalkyl Sc species generated in situ from protonolysis of the Sc dialkyl. Subsequently, complex 9 was directly applied as a catalyst for the reaction of N,N-dimethylaniline with 1-octene under standard conditions (Scheme 2, 10 mol% catalyst loading, toluene, 120 °C). Again, it selectively gave a branched C(sp3)–H alkylation product 7aa in 73% isolated yield, suggesting that the cationic o-dimethylaminophenyl scandium complex 9 behaved as a catalytic intermediate in alkylation reactions. In comparison, the treatment of the Sc complex 1 with [PhNHMe2][B(C6F5)4] afforded an analogous product 10,16 which showed neglected activity towards the reaction of N,N-dimethylaniline with 1-octene.
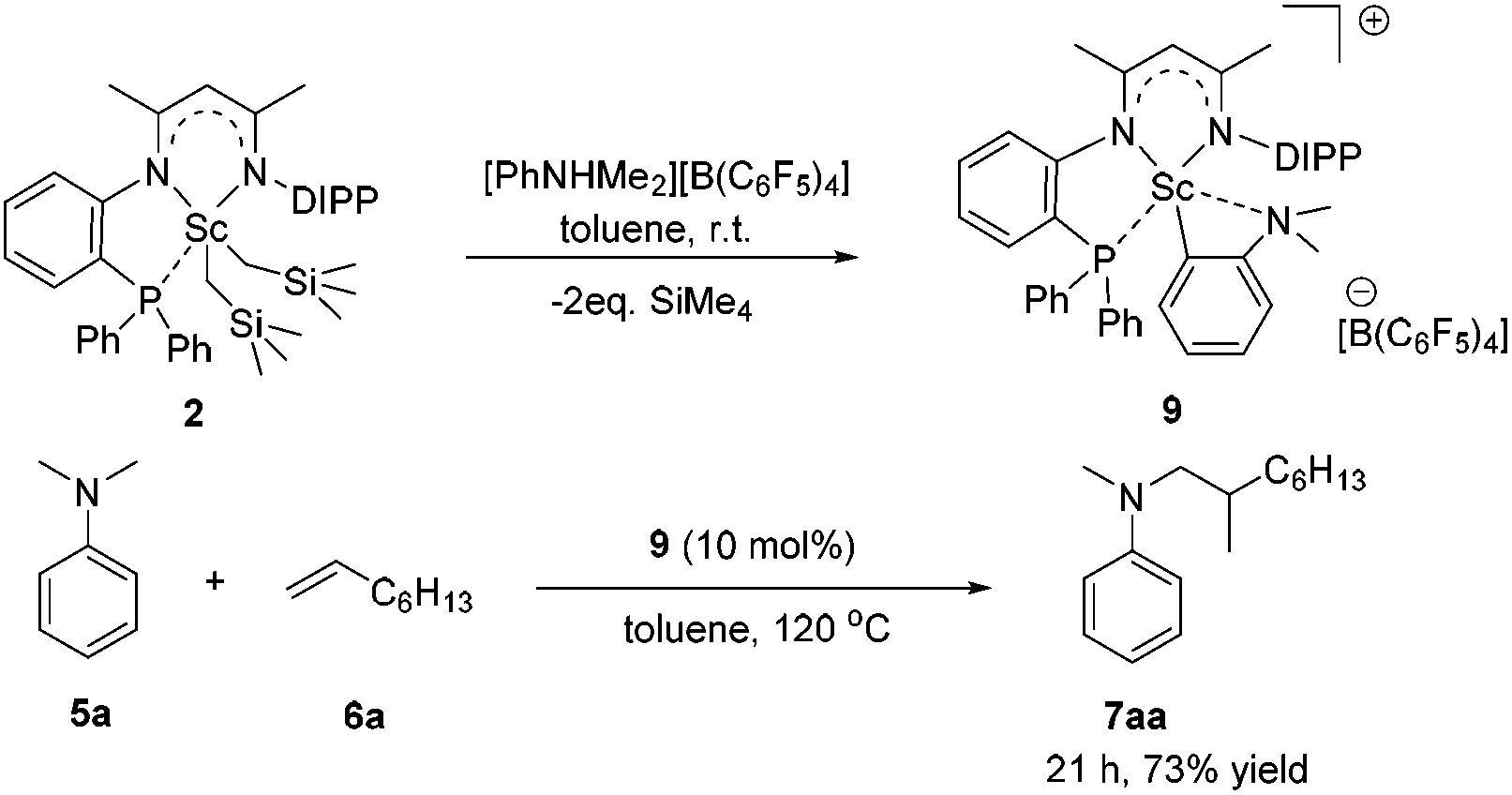 | ||
| Scheme 2 Synthesis of complex 9 and its application in a C–H alkylation reaction. | ||
The intuitive result of the reaction between 9 and alkene is the insertion of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C into the Sc–C(sp2) bond, which will lead to aryl-functionalization rather than the observed alkyl-functionalization. To shed light on the mechanism of the anti-intuitive and highly intriguing reactions, we conducted deuterium labelling experiments to provide more information. The reaction of N,N-dimethylaniline-d5 (5a-d5) with allyltrimethylsilane (6i) catalyzed by 2/[PhNHMe2][B(C6F5)4] gave the alkylation product 7ai-d5, which contained only 22% deuterium incorporation at the ortho position (Scheme 3a). Compound 7di-d6 generated by the treatment of N,N-(dimethyl-d6)-p-toluidine (5d-d6) with 6i under similar conditions showed 35% deuterium incorporation into the ortho position of the arene (Scheme 3b). In addition, the intermolecular competition experiment of 5d/5d-d6 with 6i showed a significant kinetic isotope effect (kH/kD = 2.96; Fig. S70†). These observations implied that the ortho C(sp2)–H activation of the phenyl ring had been undoubtedly involved in this scandium-catalyzed C(sp3)–H functionalization reaction.11a
C into the Sc–C(sp2) bond, which will lead to aryl-functionalization rather than the observed alkyl-functionalization. To shed light on the mechanism of the anti-intuitive and highly intriguing reactions, we conducted deuterium labelling experiments to provide more information. The reaction of N,N-dimethylaniline-d5 (5a-d5) with allyltrimethylsilane (6i) catalyzed by 2/[PhNHMe2][B(C6F5)4] gave the alkylation product 7ai-d5, which contained only 22% deuterium incorporation at the ortho position (Scheme 3a). Compound 7di-d6 generated by the treatment of N,N-(dimethyl-d6)-p-toluidine (5d-d6) with 6i under similar conditions showed 35% deuterium incorporation into the ortho position of the arene (Scheme 3b). In addition, the intermolecular competition experiment of 5d/5d-d6 with 6i showed a significant kinetic isotope effect (kH/kD = 2.96; Fig. S70†). These observations implied that the ortho C(sp2)–H activation of the phenyl ring had been undoubtedly involved in this scandium-catalyzed C(sp3)–H functionalization reaction.11a
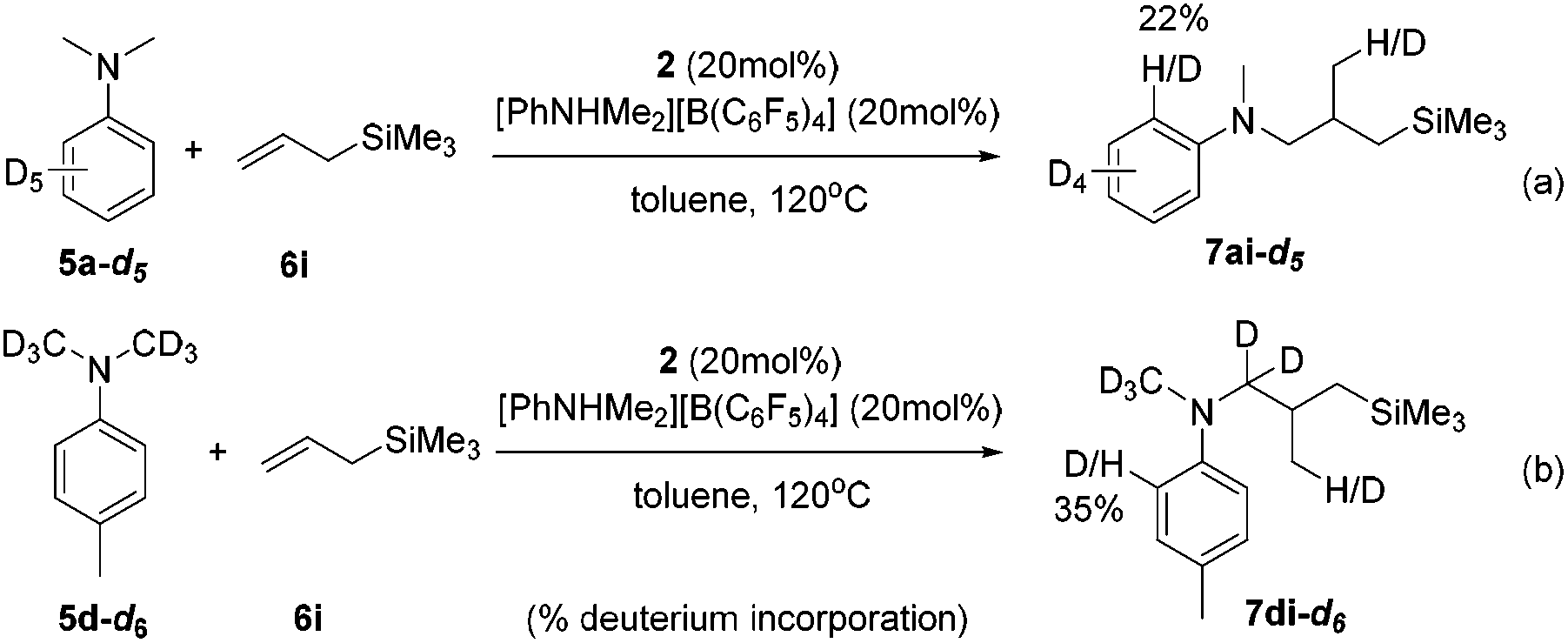 | ||
| Scheme 3 Deuterium labelling experiments of C–H alkylation reactions. | ||
Based on the above preliminary investigation, we proposed a plausible mechanistic framework for the C(sp3)–H alkylation reaction as shown in Scheme 4. The reaction of complex 2 with [PhNHMe2][B(C6F5)4] affords the cationic Sc complex 9 with liberation of SiMe4 as depicted in Scheme 4, which then might undergo intramolecular H migration upon the coordination of the CC double bond of the terminal alkene to the metal center to give the intermediate A21 rather than intuitive Sc–C(sp2) insertion. The subsequent 1,2-insertion of the coordinated alkene into the Sc–C(sp3) bond leads to the formation of the five-membered azametallacyclic complex B, which is responsible for the formation of the final branched alkylation product 7.
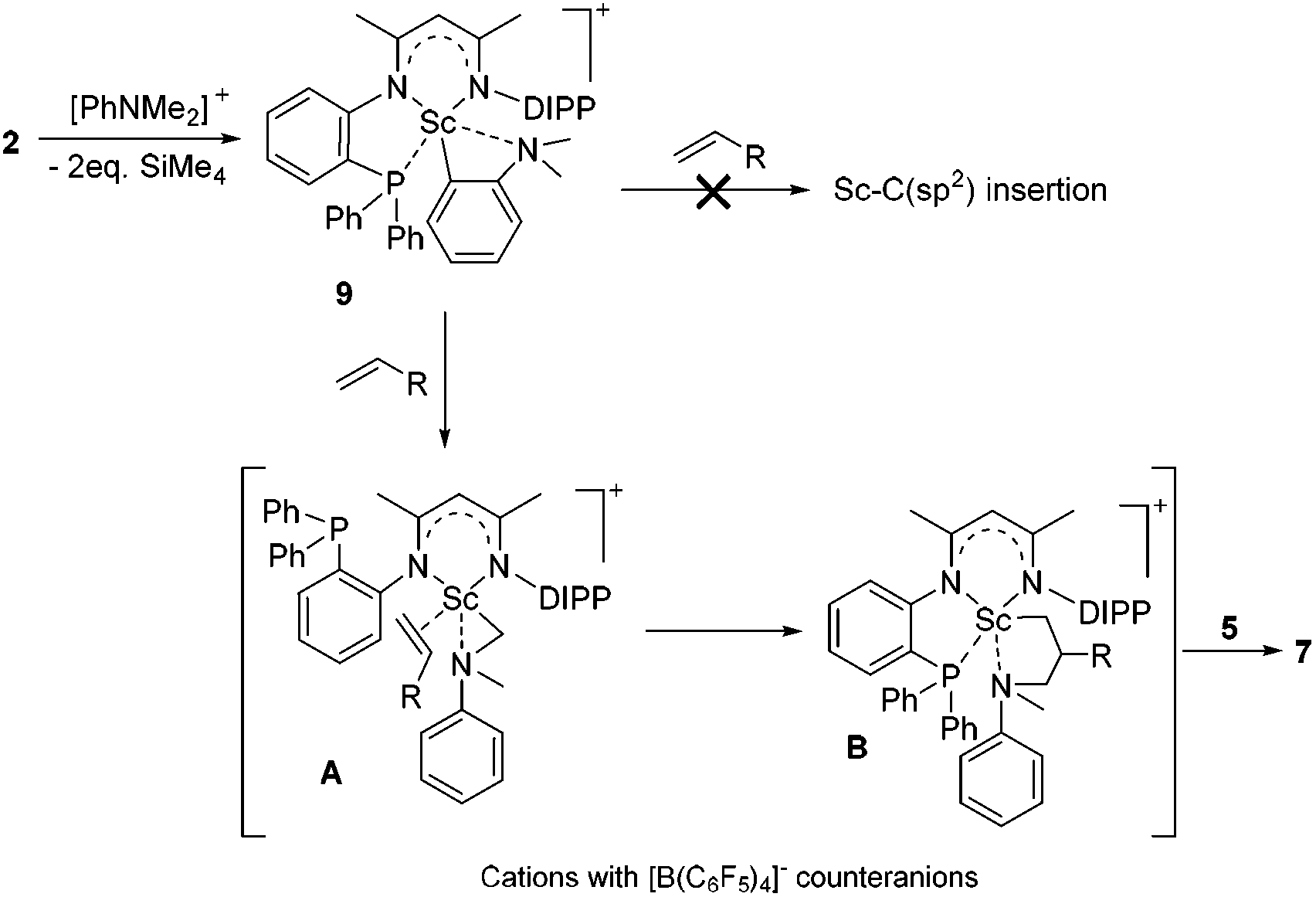 | ||
| Scheme 4 Plausible mechanistic framework for Sc-catalyzed C(sp3)–H alkylation. | ||
Conclusions
In summary, a scandium dialkyl complex stabilized by a new tridentate NNP ligand framework was synthesized and characterized. In the presence of [PhNHMe2][B(C6F5)4], the scandium dialkyl promoted the efficient and selective C(sp3)–H addition of N,N-dimethyl anilines to the CC double bonds of alkenes in the formation of tertiary aniline products with branched alkyl substituents. The work presented herein represents the first example of the catalytic C(sp3)–H addition of aromatic tertiary amines to alkenes affording branched products. The isolation of the catalytic intermediate, together with deuterium labelling experiments, revealed that the switch of ortho-C(sp2)–H activation to α-C(sp3)–H activation of the aniline substrate may be involved in this anti-intuitive and intriguing reaction. The design and choice of the ligand scaffold and rare-earth metals are crucial for tuning the reaction selectivity;22 and further comprehensive investigations are underway in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21502132), the Natural Science Foundation of Jiangsu Province (Grant No. BK20150316), the Jiangsu Specially Appointed Professor Plan, and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).Notes and references
-
(a) R. Hili and A. K. Yudin, Nat. Chem. Biol., 2006, 2, 284 CrossRef CAS PubMed
; (b) J. F. Hartwig, Acc. Chem. Res., 2008, 41, 1534 CrossRef CAS PubMed
-
(a) S. Hong and T. J. Marks, Acc. Chem. Res., 2004, 37, 673 CrossRef CAS PubMed
-
(a) A. F. Abdel-Magid, K. G. Carson, B. D. Harris, C. A. Maryanoff and R. D. Shah, J. Org. Chem., 1996, 61, 3849 CrossRef CAS PubMed
- For selected reviews, see:
(a) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed
-
(a) Z. Li, D. S. Bohle and C.-J. Li, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8928 CrossRef CAS PubMed
- H. M. L. Davies and Q. Jin, Org. Lett., 2004, 6, 1769 CrossRef CAS PubMed
-
(a) F. Kakiuchi, Top. Organomet. Chem., 2007, 24, 1 CrossRef CAS
-
(a) T. C. Boorman and I. Larrosa, Chem. Soc. Rev., 2011, 40, 1910 RSC
-
(a) J.-J. Brunet, D. Neibecker and K. Philippot, J. Chem. Soc., Chem. Commun., 1992, 1215 RSC
- G. Song, G. Luo, J. Oyamada, Y. Luo and Z. Hou, Chem. Sci., 2016, 7, 5265 RSC
-
(a) S. B. Herzon and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 6690 CrossRef CAS PubMed
-
(a) R. Kubiak, I. Prochnow and S. Doye, Angew. Chem., Int. Ed., 2009, 48, 1153 CrossRef CAS PubMed
- With the assistance of pyridine directing groups, ruthenium- or iridium-catalyzed α-C(sp3)–H alkylation of cyclic tertiary amines with alkenes affording linear products were reported. For selected examples, see:
(a) N. Chatani, T. Asaumi, S. Yorimitsu, T. Ikeda, F. Kakiuchi and S. Murai, J. Am. Chem. Soc., 2001, 123, 10935 CrossRef CAS PubMed
- Very recently, Hou and co-workers reported the first example of the C–H addition of aliphatic tertiary amines to alkenes in the formation of linear or branched products catalyzed by a homoleptic scandium trialkyl complex in combination with a borate. See: A. E. Nako, J. Oyamada, M. Nishiura and Z. Hou, Chem. Sci., 2016, 7, 6429 RSC
- P. Xu, Y. Yao and X. Xu, Chem. – Eur. J., 2017, 23, 1263 CrossRef CAS PubMed
- For preparation and characterization, see the ESI.†.
- The Sc-complex 4 was prepared according to the literature, see: P. G. Hayes, W. E. Piers, L. W. M. Lee, L. K. Knight, M. Parvez, M. R. J. Elsegood and W. Clegg, Organometallics, 2001, 20, 2533 CrossRef CAS
- Both neutral complex 2 or the [PhNHMe2][B(C6F5)4] reagent alone didn't show any activity under our typical reaction conditions.
- M. Nishiura, F. Guo and Z. Hou, Acc. Chem. Res., 2015, 48, 2209 CrossRef CAS PubMed
- For an example of the polymerization of styrene catalyzed by a cationic scandium system, see: Y. Luo, J. Baldamus and Z. Hou, J. Am. Chem. Soc., 2004, 126, 13910 CrossRef CAS PubMed
- Formation of A by intermolecular N-methyl C(sp3)–H activation of N,N-dimethylaniline cannot be ruled out at this stage.
- K. M. Engle and J.-Q. Yu, J. Org. Chem., 2013, 78, 8927 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, compound characterization, and X-ray crystallographic data for complex 2. CCDC 1568346. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qo00718c |
| This journal is © the Partner Organisations 2018 |