DOI:
10.1039/C8QM00220G
(Research Article)
Mater. Chem. Front., 2018,
2, 1568-1573
A supramolecular hyperbranched polymer with multi-responsiveness constructed by pillar[5]arene-based host–guest recognition and its application in the breath figure method†
Received
9th May 2018
, Accepted 18th June 2018
First published on 19th June 2018
Abstract
In this work, a disulfide-bridged bispillar[5]arene (A2) and tritopic neutral guest molecule (B3) were synthesized and used to construct supramolecular hyperbranched polymers (SHPs) utilizing pillar[5]arene-based host–guest interactions. In particular, a supramolecular gel with interesting reversible gel–sol transition in response to a temperature change and redox owing to the dynamic host–guest complexation and the characteristics of the disulfide bond can be obtained via the self-assembly behavior of the SHPs under high concentration. Moreover, honeycomb-patterned films can be successfully prepared by utilizing the breath figure method.
Introduction
Supramolecular polymers, which jointly have the advantages of supramolecular chemistry and polymeric materials, not only own traditional polymeric characteristics, but also possess novel structures and functions, making them exceptional candidates for materials chemistry.1,2 Moreover, the reversible and tunable nature of noncovalent interactions endows supramolecular polymers with some excellent properties, such as responsiveness to environmental stimuli,3–5 self-healing,6–8 recycling9,10 and degradability.11,12 Until now, diverse supramolecular polymers have been constructed by noncovalent interactions, such as host–guest interactions,13–15 hydrogen bonds,16–19 metal coordination20 and π–π stacks.21 Among them, supramolecular hyperbranched polymers (SHPs), which can be constructed through direct and indirect methodologies,22 have attracted great interest due to their unique chemical and physical properties and potential applications in many fields such as biochemistry23 and materials science.24
Host–guest interactions, as a kind of crucial noncovalent interaction to design supramolecular polymers, exhibit high efficiencies and distinct properties in constructing supramolecular polymer recognition motifs between macrocyclic hosts and the corresponding guests. Pillar[n]arenes,25–35 as an emerging macrocyclic host next to cyclodextrins,36,37 crown ethers,38,39 calixarenes40 and cucurbiturils,41,42 are composed of n hydroquinone units held together by methylene bridges linking the para positions in cyclic arrays. The efficient recognition capabilities and easy functionalization properties endow pillar[n]arenes with a wide range of applications in supramolecular polymers,43,44 drug delivery systems,45 sensors,46 green catalysis,47 and so on. However, the multi-responsive SHPs based on the pillar[n]arenes recognition system have rarely been reported. Herein, we constructed a SHP with thermal and redox responsiveness by the host–guest interactions between the disulfide-bridged bispillar[5]arene (A2) and tritopic neutral guest (B3) (Scheme 1). It is noteworthy that dithiothreitol (DTT) can reduce the disulfide bond into two thiols quickly via an immediate and mild process, and the regeneration of the disulfide bond can be controlled by the oxidation of iodine (I2), which endows the SHPs with efficient redox-responsiveness. Meanwhile, the host–guest interactions are quite sensitive to temperature, resulting in the thermal responsiveness of the SHPs. Moreover, the SHPs can self-assemble into a supramolecular gel with reversible gel–sol transition in response to a temperature change and redox under high concentration. Interestingly, the regeneration of the disulfide bond was realized by adding I2 as an oxidizing agent. After adding moderate I2 into the sol, an aubergine supramolecular gel was obtained. However, the gel can be transformed into an aubergine solution after the addition of excess I2. This characteristic is novel in supramolecular chemistry. Furthermore, the SHPs can be used to construct honeycomb-patterned films utilizing the breath figure method, which possesses potential applications in degradable materials by redox due to the characteristic of the disulfide bond. This work will be helpful in designing supramolecular hyperbranched polymers, supramolecular gels and 2D order materials, which may contribute to the development of their application in self-healing materials, absorption and separation.
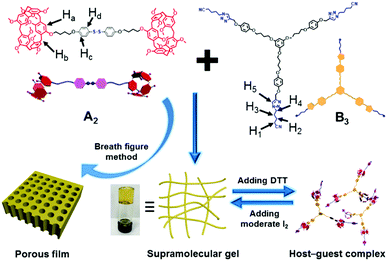 |
| | Scheme 1 Chemical structures of A2 and B3, and a cartoon representation of the formation of the supramolecular gel and porous film. | |
Results and discussion
Design and synthesis of host molecule A2 and guest molecule B3
In a previous report,48 Yang's group has synthesized a bispillararene connected by a disulfide bond by the oxidation of two thiols. However, this method is relatively tedious to prepare thiol-pillar[5]arene. Here, we constructed disulfide-bridged phenol by the oxidation of 4-hydroxythiophenol with high yield, and then the disulfide-bridged bispillar[5]arene was obtained by the substitution reaction between compound 1 and 2. On the other hand, Li's group has demonstrated a neutral guest bearing a cyano site and a triazole site at its ends,49 which is an effective guest for pillar[5]arene with an association constant of 104 M−1 in the case of an alkylated derivative. In view of this and as a part of our research effort, we prepared neutral guest B3, which has three cyano sites and triazole sites at the end of the molecule via a substitution reaction and click reaction.
Host–guest complexation studies
First, an attempt to study the interaction between the disulfide-bridged bispillar[5]arene A2 and neutral guest B3 was carried out by 1H NMR characterization in CDCl3 (Fig. 1). The resulting 1H NMR spectrum showed obvious upfield shifts of the signals of protons H1, H2, H3, H4 and H5 from molecule B3 while the signals of protons of Ha and Hb on A2 shifted downfield, indicating that the cyanoalkyl groups of B3 were encapsulated into the cavity of pillararene and further demonstrating the complexation between disulfide-bridged bispillar[5]arene A2 and neutral guest B3. In addition, from the 2D NOESY NMR spectrum of the solution of A2 and B3, NOE correlation signals were observed between the Ha and Hb protons of bispillar[5]arene A2 and H1–5 of guest molecule B3, which convincingly verified the formation of the host–guest complex (Fig. S9, ESI†).
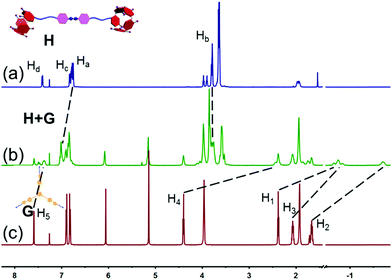 |
| | Fig. 1 Partial 1H NMR spectra (600 MHz, 298 K) in CDCl3: (a) 6.00 mM A2; (b) 6.00 mM A2 and 4.00 mM B3; (c) 4.00 mM B3. | |
Characterization of the SHPs
A supramolecular hyperbranched polymer was expected to be obtained via host–guest interactions in CHCl3. The concentration-dependent 1H NMR spectra of the A2–B3 complexes provided vital insights into the self-assembly behavior. As shown in Fig. 2a, the 1H NMR spectra of the A2–B3 complexes at concentrations of A2 in the range of 1.00–50.0 mM were tanglesome due to the slow-exchange host–guest interactions on the NMR time scale. At a low concentration of A2 (1.00 mM), the signals of protons H4 and H3 were split into two sets of peaks, corresponding to complexed branched oligomers (H4om and H3om) and uncomplexed molecules (H3uc), proving the slow-exchange binding between A2 and B3. With the monomer concentration increasing, the signals of protons H1, H2 and H3 of the cyanoalkyl groups shifted downfield. In addition, all of the main peaks broadened markedly, which gave clear evidence for the formation of supramolecular polymers.
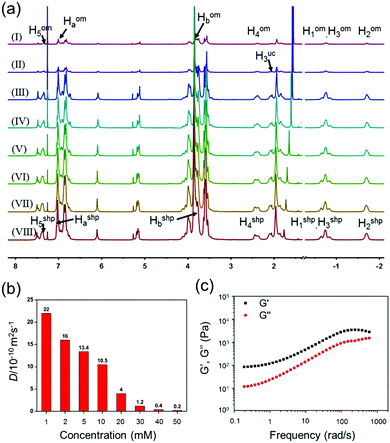 |
| | Fig. 2 (a) Partial 1H NMR spectra (600 MHz, CDCl3, 298 K) of mixtures of a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio of A2 and B3 at different concentrations of A2: (I) 1.00 mM, (II) 2.00 mM, (III) 5.00 mM, (IV) 10.0 mM, (V) 20.0 mM, (VI) 30.0 mM, (VII) 40.0 mM, (VIII) 50.0 mM. Peaks of uncomplexed monomers, oligomers, and supramolecular hyperbranched polymers, are designated as uc, om, and shp, respectively; (b) concentration dependence of the diffusion coefficient D (600 MHz, 298 K) in CDCl3 of mixtures of a 3:2 molar ratio of A2 and B3 at different A2 concentrations: 1.00 mM, 2.00 mM, 5.00 mM, 10.0 mM, 20.0 mM, 30.0 mM, 40.0 mM, 50.0 mM; (c) rheological results of the supramolecular gel constructed by 75.0 mM A2 and 50.0 mM B3 in CHCl3. :2 molar ratio of A2 and B3 at different concentrations of A2: (I) 1.00 mM, (II) 2.00 mM, (III) 5.00 mM, (IV) 10.0 mM, (V) 20.0 mM, (VI) 30.0 mM, (VII) 40.0 mM, (VIII) 50.0 mM. Peaks of uncomplexed monomers, oligomers, and supramolecular hyperbranched polymers, are designated as uc, om, and shp, respectively; (b) concentration dependence of the diffusion coefficient D (600 MHz, 298 K) in CDCl3 of mixtures of a 3:2 molar ratio of A2 and B3 at different A2 concentrations: 1.00 mM, 2.00 mM, 5.00 mM, 10.0 mM, 20.0 mM, 30.0 mM, 40.0 mM, 50.0 mM; (c) rheological results of the supramolecular gel constructed by 75.0 mM A2 and 50.0 mM B3 in CHCl3. | |
In addition, 2D diffusion-ordered 1H NMR spectroscopy (DOSY) experiments were performed to provide straightforward evidence for the formation of SHPs. As shown in Fig. 2b, the measured weight-average diffusion coefficient D values of the A2–B3 complexes decreased from 22.0 × 10−10 m2 s−1 to 0.2 × 10−10 m2 s−1 with an increase in the concentration of A2 (with 2/3 equiv. B3) from 1.00 mM to 50.0 mM, indicating that the high-molecular-weight polymeric species were successfully formed. Interestingly, a yellowish transparent supramolecular gel can be obtained upon increasing the concentration of the monomers (75.0 mM A2 and 50.0 mM B3).
The characterization of the supramolecular gel
Rheological characterization is an important analytical tool to characterize the supramolecular gel because it can display the material properties directly. Therefore, an oscillatory sweep experiment was carried out to obtain the rheological properties of the supramolecular gel (75 mM A2 with 2/3 equiv. B3, Fig. 2c). The result of the rheological experiment showed the frequency dependence of the storage modulus G′ and loss modulus G′′ of the supramolecular gel, and G′ was larger than G′′ over a wide frequency range of 0.1–100 rad s−1, indicating that the supramolecular gel was stable. Moreover, the morphology of the supramolecular gel was characterized by transmission electron microscopy (TEM) and scanning electron microscopy (SEM). As shown in Fig. 3, a three-dimensional network structure could be observed clearly.
 |
| | Fig. 3 (a), (b) TEM images and (c), (d) SEM images of the supramolecular polymer network of the mixtures of a 3:2 molar ratio of A2 and B3 at 20.0 mM of A2. | |
Supramolecular gels that can exhibit a response to external stimuli are always attractive. It's well known that host–guest interactions are quite sensitive to temperature. The temperature-variant NMR experiments of A2–B3 complexes showed the reduction of the intensity of the peaks of H1, H2, and H3 along with the increase of temperature from 298 K to 328 K (Fig. S16, ESI†), which indicated that the host–guest interaction decreased. Therefore, as shown in Fig. 4a, heating and cooling can result in a reversible gel–sol transition. The disulfide bond has gained extensive attention because of its biological activities and potential reactivity.50 Furthermore, the cleavage and formation of the disulfide bond can be controlled by redox. To examine the redox-responsiveness of compound A2 in more detail, cyclic voltammetry was conducted in a solution of tetrabutylammonium hexafluorophosphate (0.10 mM in CHCl3) (Fig. S17a, ESI†). Obvious peaks in the cyclic voltammetry curves were observed, which indicated that the cleavage and formation of the disulfide bond can be controlled by redox. Moreover, DTT, as one of the most used reductants, was selected to reduce the disulfide bonds under mild conditions. As shown in Fig. 4b, upon the addition of 1.00 equiv. of DTT into the supramolecular gel, the gel gradually collapsed within 2 hours, resulting in a gel–sol transition. Furthermore, I2 is widely used for disulfide bond formation reactions with a rapid rate and high yield in peptide synthesis. Therefore, here we chose I2 as an oxidizing agent to realize the regeneration of the disulfide bond. Upon the addition of moderate I2 into the sol, an aubergine supramolecular gel was ultimately obtained within 4 hours. Interestingly, after adding excess I2 into the aubergine supramolecular gel, the gel was destroyed and transformed into an aubergine solution. According to the previous work, I2 can locate in the cavity of pillar[5]arene which destroyed the host–guest interaction between pillar[5]arene and the guest molecule.46,511H NMR study was further used to investigate the mechanism in CDCl3 (Fig. S17b, ESI†). The DMP5 was selected as a model host molecule, and the 1H NMR spectra of DMP5 and DMP5 with excess I2 are shown in Fig. S17b (ESI†). The resulting 1H NMR spectra showed that the signals of the protons on DMP5 shifted downfield, indicating the existence of the interactions between the pillararene and I2, which could hinder the host–guest interaction between pillararene and other guest molecules. Moreover, the aubergine solution could be transformed into a supramolecular gel by sublimation in air, which endowed the material with reversible stimuli-responsiveness (Fig. 4c).
 |
| | Fig. 4 The gel–sol transitions of the supramolecular gel triggered by (a) heating and cooling, (b) redox, and (c) adding excess I2 and sublimation. | |
Honeycomb-patterned films fabricated by the breath figure method
Inspired by the three-dimensional network structure of SHPs (Fig. 3), the obtained SHPs were tested to determine whether they could be used to fabricate potential topological supramolecular materials such as 2D honeycomb-patterned films, which have potential applications in the fields of separation,52 drug release53 and catalysis.54 As shown in Fig. 5, an ordered honeycomb film was prepared using the breath figure method. A 7.5 wt% concentration of SHPs in CHCl3 was used to prepare the honeycomb-patterned structure, the average pore diameter of which was about 1.0–1.5 μm by SEM analysis. Above all, the disulfide bond can be destroyed by the addition of DTT. Therefore, DTT can be used to control the formation of the honeycomb-patterned films. After adding 1.0 equiv. DTT into the solution of SHPs (7.5 wt%) in CHCl3, irregular particles were formed (Fig. 5c and d). This is direct evidence that the honeycomb-patterned films were fabricated based on SHPs instead of small molecules, and this characteristic endows the honeycomb-patterned films with degradable properties.
 |
| | Fig. 5 (a) Digital photo of the honeycomb-patterned films made from a solution of A2–B3 complexes in CHCl3 at the concentration of 7.5 wt% using the BF method; (b) SEM of the honeycomb-patterned films. (c and d) SEM images of films made from a solution of A2–B3 complexes in CHCl3 at the concentration of 7.5 wt% after adding 1.0 equiv. DTT using the BF method. | |
Conclusions
In summary, we have designed and synthesized a disulfide-bridged bispillar[5]arene A2 and a tritopic neutral guest molecule B3, which can self-assemble into supramolecular hyprebranched polymers, and the supramolecular hyprebranched polymers can form a supramolecular gel at a high concentration of the monomers based on the host–guest interactions between pillar[5]arenes and the neutral guests. The supramolecular polymer gel possessed multi-responsiveness and the reversible gel–sol transitions of the supramolecular gel can be triggered by heating, cooling, redox, adding excess I2 and sublimation, which can facilitate its application as a smart material. Moreover, the resulting SHPs can be employed to prepare honeycomb-patterned films using the breath figure method. Furthermore, the honeycomb-patterned films have potential applications in degradable materials by redox due to the characteristic of the disulfide bond. This work will be helpful in designing supramolecular hyperbranched polymers, supramolecular gels and 2D ordered materials, which may contribute to the development of their applications in self-healing materials, absorption and separation.
Experimental
Materials and instrumentation
All reagents were commercially available and used as supplied without further purification. Compounds 1,552,563,57 and 631 were synthesized according to the previous work. 1H NMR spectra, 13C NMR, NOESY and DOSY NMR spectra were recorded with an Agilent DD2-600 with the use of the deuterated solvent as the lock and the residual solvent or TMS as the internal reference. Scanning electron microscopy investigations were carried out on a JEOL 6390LV instrument. SEM samples were prepared via the vacuum freeze-drying methodology. Transmission electron microscopic investigations were carried out on a HITACHI HT-7700 instrument. High-resolution mass spectrometric experiments were performed using a Bruker 7 Tesla FT-ICR mass spectrometer equipped with an electrospray source (Billerica, MA, USA). The disc-shaped gels with thickness of 1 mm and diameter of 20 mm were adhered to the plates and surrounded by silicone oil. Frequency sweeps were performed on the samples with a strain amplitude of 0.05%.
Synthesis of A2
The synthetic route of A2 is shown in the Scheme S1 (ESI†). A mixture of compound 1 (1.00 g, 1.41 mmol), 2 (0.14 g, 0.56 mmol), and K2CO3 (0.78 g, 0.56 mmol) in CH3CN (75 mL) was stirred at reflux for 48 hours. After the solid was filtered off, the solvent was concentrated by rotary evaporation. The crude product was dissolved in CH2Cl2 (100 mL) and washed three times with H2O (50 mL). The organic layer was dried over anhydrous Na2SO4 and concentrated to afford a jacinth solid, which was further purified by column chromatography using ethyl acetate/petroleum ether (1:2). The fractions containing the product were concentrated to give A2 as a white solid (0.63 g, 68%). The 1H NMR spectrum of A2 is shown in Fig. S1 (ESI†). 1H NMR spectrum of A2 (600 MHz, 298 K) in CDCl3δ (ppm): 7.40–7.42 (d, J = 12 Hz, 4H), 6.82–6.83 (d, J = 6 Hz, 4H), 6.75–6.77 (m, 20H), 3.96–3.99 (t, J = 6 Hz, 4H), 3.89–3.91 (t, J = 6 Hz, 4H), 3.79–3.81 (m, 20H), 3.63–3.66 (m, 54H), 1.98 (m, 8H). The 13C NMR spectrum of A2 is shown in Fig. S2 (ESI†). 13C NMR spectrum of A2 (150 MHz, 298 K) in CDCl3δ (ppm): 159.5, 151.0, 150.1, 132.8, 128.5, 115.2, 114.2, 68.1, 67.8, 56.0, 29.9, 26.3. HR ESI-MS: m/z calcd for [M + H+] C108H119O22S2+, 1831.76; found 1831.77, error 5 ppm.
Synthesis of compound 4
The synthetic route of B3 is shown in the Scheme S2 (ESI†). A solution of compound 3 (5.29 g, 10.0 mmol), compound 5 (5.28 g, 40.0 mmol) and K2CO3 (13.8 g, 100 mmol) in CH3CN (250 mL) was stirred at reflux for 48 hours. After the solid was filtered off, the mixture was evaporated to afford the crude product, which was isolated by flash column chromatography using ethyl acetate/petroleum ether (1:10) to give 4 as a white solid (7.25 g, 78%). The 1H NMR spectrum of 4 is shown in Fig. S4 (ESI†). 1H NMR spectrum of 4 (600 MHz, 298 K) in CDCl3δ (ppm): 6.76–6.86 (m, 12H), 5.99 (s, 3H), 4.56 (d, J = 6 Hz, 6H), 3.90 (s, 12H), 2.43 (t, J = 3 Hz, 3H), 1.87 (m, 12H). The 13C NMR spectrum of 4 is shown in Fig. S5 (ESI†). 13C NMR spectrum of 4 (150 MHz, 298 K) in CDCl3δ (ppm): 159.8, 152.8, 150.6, 115.1, 114.3, 92.8, 77.9, 74.3, 66.9, 55.6, 25.0.
Synthesis of guest molecule B3
A solution of compound 4 (6.60 g, 9.0 mmol), compound 6 (5.1 g, 41.1 mmol), CuSO4·5H2O (2.27 g, 9.0 mmol) and sodium ascorbate (2.67 g, 18 mmol) in 80 mL DMF was stirred at 90 °C for 24 hours. After the reaction finished, the crude product was dissolved in CH2Cl2 (400 mL) and washed three times with H2O (200 mL). The organic layer was dried over anhydrous Na2SO4 and concentrated to afford a grayish yellow solid, which was further purified by recrystallization in moderate CH3CN (5.35 g, 54%). The 1H NMR spectrum of B3 is shown in Fig. S7 (ESI†). 1H NMR spectrum of B3 (600 MHz, 298 K) in CDCl3δ (ppm): 7.59 (s, 3 H), 6.81–6.90 (m, 12H), 6.06 (s, 3H), 5.14 (s, 3H), 6.22 (t, J = 6 Hz, 3H), 3.96 (s, 12H), 2.39 (t, J = 6 Hz, 6H), 2.09 (m, 6H), 1.92 (s, 12H), 5.99 (m, 6H). The 13C NMR spectrum of B3 is shown in Fig. S8 (ESI†). 13C NMR spectrum of B3 (150 MHz, 298 K) in CDCl3δ (ppm): 160.9, 153.7, 152.4, 144.9, 122.7, 119.0, 116.0, 115.6, 94.1, 68.1, 67.6, 62.8, 49.3, 29.1, 26.2, 26.0, 22.4, 16.8. HR ESI-MS: m/z calcd for [2M + H+ + K+] C120H145N24O18K2+, 1124.54; found 1124.51, error −3 ppm.
Fabrication of honeycomb-patterned films by the breath figure method
A mixture of a 3:2 molar ratio of A2 and B3 was dissolved in chloroform to obtain a solution of SHPs at a concentration of 7.5 wt%. Next, about 50 μL of the polymer solution was dripped on the treated PET substrate by a syringe. During this process, a steady airflow of moist nitrogen was maintained through the surface of the polymer solution. The temperature could be decreased due to the evaporation of chloroform, which resulted in the condensation of water vapor on the surface of the solution. Subsequently, chloroform and water was evaporated conjointly, and an ordered porous structure was finally formed on the cover of the polymer membrane.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Fundamental Research Funds for the Central Universities.
Notes and references
- T. F. A. De. Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687 CrossRef PubMed.
- S. Dong, B. Zheng, F. Wang and F. Huang, Acc. Chem. Res., 2014, 47, 1982 CrossRef PubMed.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14 CrossRef PubMed.
- X. Ma and H. Tian, Acc. Chem. Res., 2014, 47, 1971 CrossRef PubMed.
- W. Xia, M. Ni, C. Yao, X. Wang, D. Chen, C. Lin, X.-Y. Hu and L. Wang, Macromolecules, 2015, 48, 4403 CrossRef.
- A. Phadke, C. Zhang, B. Arman, C.-C. Hsu, R. A. Mashelkar, A. K. Lele, M. J. Tauber, G. Arya and S. Varghese, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4383 CrossRef PubMed.
- J. W. Steed, Chem. Soc. Rev., 2010, 39, 3686 RSC.
- T. Kakuta, Y. Takashima, M. Nakahata, M. Otsubo, H. Yamaguchi and A. Harada, Adv. Mater., 2013, 25, 2849 CrossRef PubMed.
- X. Li, J. Fei, Y. Xu, D. Li, T. Yuan, G. Li, C. Wang and J. Li, Angew. Chem., Int. Ed., 2018, 57, 1903 CrossRef PubMed.
- M. Abbasi, J. Chin. Chem. Soc., 2017, 64, 896 CrossRef.
- Y. Kohsaka, K. Nakazono, Y. Koyama, S. Asai and T. Takata, Angew. Chem., Int. Ed., 2011, 50, 4872 CrossRef PubMed.
- Q. Wang, M. Cheng, L. Tian, Q. Fan and J. Jiang, Polym. Chem., 2017, 8, 6058 RSC.
- A. Harada, Y. Takashima and M. Nakahata, Acc. Chem. Res., 2014, 47, 2128 CrossRef PubMed.
- H. Yang, B. Yuan and X. Zhang, Acc. Chem. Res., 2014, 47, 2106 CrossRef PubMed.
- G. Yu, Z. Yang, X. Fu, B. C. Yung, J. Yang, Z. Mao, L. Shao, B. Hua, Y. Liu, F. Zhang, Q. Fan, S. Wang, O. Jacobson, A. Jin, C. Gao, X. Tang, F. Huang and X. Chen, Nat. Commun., 2018, 9, 766 CrossRef PubMed.
- T. Park and S. C. Zimmerman, J. Am. Chem. Soc., 2006, 128, 13986 CrossRef PubMed.
- Y. Ma, S. V. Kolotuchi and S. C. Zimmerman, J. Am. Chem. Soc., 2002, 124, 13757 CrossRef PubMed.
- X. Yan, Z. Liu, Q. Zhang, J. Lopez, H. Wang, H.-C. Wu, S. Niu, H. Yan, S. Wang, T. Lei, J. Li, D. Qi, P. Huang, J. Huang, Y. Zhang, Y. Wang, G. Li, J. B.-H. Tok, X. Chen and Z. Bao, J. Am. Chem. Soc., 2018, 140, 5280 CrossRef PubMed.
- Z. Gao, Y. Han, S. Chen, Z. Li, H. Tong and F. Wang, ACS Macro Lett., 2017, 6, 541 CrossRef.
- F. Peng, G. Li, X. Liu, S. Wu and Z. Tong, J. Am. Chem. Soc., 2008, 130, 16166 CrossRef PubMed.
- I. Welterlich and B. Tieke, Macromolecules, 2011, 44, 4194 CrossRef.
- W. Tian, X. Li and J. Wang, Chem. Commun., 2017, 53, 2531 RSC.
- G. Fernández, E. M. Pérez, L. Sánche and N. Martín, J. Am. Chem. Soc., 2008, 130, 2410 CrossRef PubMed.
- R. Gu, J. Yao, X. Fu, W. Zhou and D.-H. Qu, Chem. Commun., 2015, 51, 5429 RSC.
- T. Ogoshi, S. Kanai, S. Fujinami, T. A. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022 CrossRef PubMed.
- D. Cao, Y. Kou, J. Liang, Z. Chen, L. Wang and H. Meier, Angew. Chem., Int. Ed., 2009, 48, 9721 CrossRef PubMed.
- T. Ogoshi, H. Kayama, D. Yamafuji, T. Aoki and T. Yamagishi, Chem. Sci., 2012, 3, 3221 RSC.
- X.-F. Ji, D.-Y. Xia, X. Yan, H. Wang and F.-H. Huang, Acta Polym. Sin., 2017, 9–18 Search PubMed.
- X.-B. Hu, L. Chen, W. Si, Y. Yu and J.-L. Hou, Chem. Commun., 2011, 47, 4694 RSC.
- Y.-M. Zhang, B. Shi, H. Li, W.-J. Qu, G.-Y. Gao, Q. Lin, H. Yao and T.-B. Wei, Polym. Chem., 2014, 5, 4722 RSC.
- Y. Liu, L. Shangguan, H. Wang, D. Xia and B. Shi, Polym. Chem., 2017, 8, 3783 RSC.
- X. Liao, L. Guo, J. Chang, S. Liu, M. Xie and G. Chen, Macromol. Rapid Commun., 2015, 36, 1492 CrossRef PubMed.
- G. Yu, J. Yang, X. Fu, Z. Wang, L. Shao, Z. Mao, Y. Liu, Z. Yang, F. Zhang, W. Fan, J. Song, Z. Zhou, C. Gao, F. Huang and X. Chen, Mater. Horiz., 2018, 5, 429 RSC.
- J.-F. Chen, Q. Lin, Y.-M. Zhang, H. Yao and T.-B. Wei, Chem. Commun., 2017, 53, 13296 RSC.
- J.-F. Chen, Q. Lin, H. Yao, Y.-M. Zhang and T.-B. Wei, Mater. Chem. Front., 2018, 2, 999 RSC.
- J. Liu, G. Chen, M. Guo and M. Jiang, Macromolecules, 2010, 43, 8086 CrossRef.
- Z.-X. Zhang, X. Liu, F. J. Xu, X. J. Loh, E.-T. Kang, K.-G. Neoh and J. Li, Macromolecules, 2008, 41, 5967 CrossRef.
- N. Yamaguchi and H. W. Gibson, Angew. Chem., Int. Ed., 1999, 38, 143 CrossRef.
- H. W. Gibson, Y. X. Shen, M. C. Bheda and C. Gong, Polymer, 2014, 55, 3202 CrossRef.
- D.-S. Guo and Y. Liu, Chem. Soc. Rev., 2012, 41, 5907 RSC.
- K. Kim, Chem. Soc. Rev., 2002, 31, 96 RSC.
- S. Ghosh and L. Isaacs, J. Am. Chem. Soc., 2010, 132, 4445 CrossRef PubMed.
- B. Shi, D. Xia and Y. Yao, Chem. Commun., 2014, 50, 13932 RSC.
- Z. Zhang, Y. Luo, J. Chen, S. Dong, Y. Yu, Z. Ma and F. Huang, Angew. Chem., Int. Ed., 2011, 50, 1397 CrossRef PubMed.
- Q. Duan, Y. Cao, Y. Li, X. Hu, T. Xiao, C. Lin, Y. Pan and L. Wang, J. Am. Chem. Soc., 2013, 135, 10542 CrossRef PubMed.
- Q. Lin, K.-P. Zhong, J.-H. Zhu, L. Ding, J.-X. Su, H. Yao, T.-B. Wei and Y.-M. Zhang, Macromolecules, 2017, 50, 7863 CrossRef.
- Y. Liu, B. Lou, L. Shangguan, J. Cai, H. Zhu and B. Shi, Macromolecules, 2018, 51, 1351 CrossRef.
- C.-L. Sun, J.-F. Xu, Y.-Z. Chen, L.-Y. Niu, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, Polym. Chem., 2016, 7, 2057 RSC.
- C. Li, K. Han, J. Li, Y. Zhang, W. Chen, Y. Yu and X. Jia, Chem. – Eur. J., 2013, 19, 11892 CrossRef PubMed.
- R. Singh and G. M. Whitesides, J. Am. Chem. Soc., 1990, 112, 1190 CrossRef.
- K. Jie, Y. Zhou, E. Li, Z. Li, R. Zhao and F. Huang, J. Am. Chem. Soc., 2017, 139, 15320 CrossRef PubMed.
- J. Zhang, Z. Meng, J. Liu, C. Schlaich, Z. Yu and X. Deng, J. Mater. Chem. A, 2017, 5, 16369 RSC.
- Y. Su, J. Dang, H. Zhang, Y. Zhang and W. Tian, Langmuir, 2017, 33, 7393 CrossRef PubMed.
- L. Zhang, L. Chen, S.-X. Liu, J. Gong, Q. Tang and Z.-M. Su, Dalton Trans., 2018, 47, 105 RSC.
- L. Liu, D. Cao, Y. Jin, H. Tao, Y. Kou and H. Meier, Org. Biomol. Chem., 2011, 9, 7007 RSC.
- F. Vibert, S. R. A. Marque, E. Bloch, S. Queyroy, M. P. Bertrand, S. Gastaldi and E. Besson, Chem. Sci., 2014, 5, 4716 RSC.
- H. Yang, Z. Ma, Z. Wang and X. Zhang, Polym. Chem., 2014, 5, 1471 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, characterization and other materials. See DOI: 10.1039/c8qm00220g |
|
| This journal is © the Partner Organisations 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *b and
Bingbing
Shi
*b and
Bingbing
Shi





