
Enhancement of the photocatalytic performance and thermal stability of an iron based metal–organic-framework functionalised by Ag/Ag3PO4
Feroz Ahmad
Sofi
and
Kowsar
Majid
 *
*
Department of Chemistry, National Institute of Technology, Srinagar, J&K-190006, India. E-mail: kowsarmajid@rediffmail.com; Tel: +919469804701
First published on 19th February 2018
Abstract
Metal–organic-frameworks have been reported as potential photocatalyst candidates due to their high surface area and semiconductive behaviour, but due to their high charge recombination rate and low visible light harvesting efficiency their use as photocatalysts has remained a challenge. A promising strategy to overcome these shortcomings is the construction of hybrid nanocomposite photocatalysts which reduce the recombination of charge carriers and simultaneously enhance the light harvesting efficiency. Herein, we report the microwave-assisted design of a novel visible light active composite material consisting of an iron-based metal–organic-framework functionalised by Ag/Ag3PO4. The resulting composite material demonstrates that both Ag3PO4 and MIL-53(Fe) show improved charge separation involving a Z-scheme internal charge transmission mechanism via ‘Ag’ nanoparticles (Ag0 NPs). The high dispersion of Ag0 and Ag3PO4 NPs and their close contact with the MIL-53(Fe) matrix help to degrade the organic pollutants effectively under visible light due to their synergistic effects. The photocatalytic degradation of a hazardous sulfonated azo dye Ponceau BS was carried out under visible light irradiation and Ag/Ag3PO4/MIL-53(Fe) was found to exhibit the highest photocatalytic performance. In addition, thermogravimetric analysis reveals that functionalization of MIL-53(Fe) by Ag/Ag3PO4/increases the thermal stability of MIL-53(Fe) which is attributed to the strengthening of an organic linker attached to iron due to the interaction of Ag/Ag3PO4 with the framework. Scavenger experiments supported the proposed Z-scheme internal charge separation. Moreover, the reusability experiments clearly indicate the high stability of the catalysts.
1. Introduction
Metal–organic-frameworks (MOFs) constructed by the self-assembly of metal ions and organic bridging ligands are a class of promising porous hybrid materials.1 MOFs have shown great potential applications due to their elegant topology, high surface area, tunable pore size and physiochemical functionality, in many fields such as gas separation and storage, heterogeneous catalysis, magnetic and electronic devices, chemical sensing and drug delivery.1–6 Furthermore, MOFs have been reported to exhibit semiconducting behaviour suggesting that these MOFs can directly harvest solar energy and can also facilitate charge transfer from organic linkers to the metal (i.e., linker-metal charge transfer, LMCT).7–11 These features enable their applications in the field of photocatalysis and thus make them a potential candidate material for application in the photocatalysis field. In the recent past, researchers have focussed on the use of MOFs for heterogeneous photocatalysis to drive solar energy conversion reactions like organic transformations, water splitting and degradation of pollutants.12–14 However, the lack of visible light response at longer wavelengths and low efficiency in charge separation has rendered these MOFs, by and large, unexplored. Some attempts have been made to tune their optical properties by modifying organic linkers which makes them highly active under visible light irradiation. For example, MIL-125(Ti) was functionalized with amino groups as reported by Gascon et al.15 However, these post synthetic modifications have been demonstrated only on MOFs formed from specific terephthalate linkers. Besides the post modification or judicious selection of bridging ligands, there are several other methods to improve the visible light photocatalytic ability of MOFs including incorporation of photoactive organic molecules such as anthracene derivatives and porphyrins.16–18In the recent past, many functional entities such as carbon materials like g-C3N4 and graphene, metal oxides/sulphides, and metal nanoparticles were incorporated into MOFs resulting in excellent MOF-based multicomponent photocatalytic materials such as TiO2@salicylaldehyde-NH2-MIL-101(Cr),19 Pt NPs@MOFs,20 graphene based MIL-53(Fe),21 and Bi2WO6@UiO66.22 These studies clearly indicate that composite materials show superior photocatalytic performance than pristine MOF materials.
Among the various types of MOFs, MIL-53(Fe), an iron-based MOF, has received increased attention from researchers. MIL-53(Fe) has been extensively researched in the fields of catalysis, hydrogen generation, gas storage, and drug delivery.23,24 It is also expected to be a promising candidate for catalytic applications in an aqueous medium, such as water splitting and water treatment, especially under visible-light irradiation. Although researchers have modified MIL-53(Fe) with various functional materials for efficient visible light driven activity, there are very limited reports on the functionalization of MIL-53(Fe) for water treatment. Furthermore, the functionalization of MIL-53(Fe) by metal nanoparticles to enhance the visible light photocatalytic activity and thermal stability has not been reported yet. Herein, we report for the first time the functionalization of an iron based metal–organic-framework, MIL-53(Fe), by incorporating a cost-effective cocatalyst, Ag–Ag3PO4.
In this work, we report a systematic study on the Ag/Ag3PO4/MIL-53(Fe) composite photocatalyst prepared by a microwave method for the degradation of a hazardous and toxic organic pollutant Ponceau BS. Ag3PO4 is a narrowband gap semiconductor with unique photooxidative ability25–27 and has been extensively used as a photocatalyst to enhance the photoactivity of many pristine materials which are otherwise photo inactive or less active. Since photocorrosion of silver containing materials is an important concern, metal nanoparticles i.e., Ag NPs, were loaded onto the surface of the Ag3PO4/MIL-53(Fe) catalyst. These Ag NPs act as cocatalysts and facilitate charge separation at the heterojunction interface via the Z-scheme internal charge transmission mechanism.28–30 The functionalization of MIL-53(Fe) by Ag/Ag3PO4 also increases the thermal stability of the MOF because it strengthens the attachment of an organic linker attached to iron due to the interaction of Ag/Ag3PO4 with the framework.
2. Experimental
2.1. Chemicals
Silver nitrate (99.0%), terephthalic acid (1,4-benzenedicarboxylic acid), ferric chloride hexahydrate (FeCl3·6H2O), di-sodium hydrogen phosphate (Na2HPO4, 98.5%), Ponceau BS (PBS), benzoquinone (BQ), absolute ethanol, tertiary butyl alcohol (TBA) and N,N-dimethylformamide (DMF, 99.8%, ACS grade, BDH) were purchased from Sigma-Aldrich and were used without further purification. Ultra-pure water was used throughout the study.2.2. Synthesis of MIL-53(Fe)
MIL-53(Fe) was synthesized by a microwave-assisted process. Generally, 0.5125 g of terephthalic acid and 0.5000 g of FeCl3·6H2O were added to 30 ml of DMF solvent and the mixture was stirred for 20 min. To this mixture, 30 mL of ethanol was added and the resulting solution was placed in a microwave synthesizer (monowave) at 100 °C for 30 min. After cooling down to 50 °C, the resulting solid was collected by centrifugation and washed with a DMF–ethanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, v/v) mixture and finally dried in a vacuum at 150 °C for 5 h.
:5, v/v) mixture and finally dried in a vacuum at 150 °C for 5 h.
2.3. Synthesis of the Ag3PO4/MIL-53(Fe) composite
The Ag3PO4@MIL-53(Fe) composite was synthesized by a simple precipitation–deposition method. Typically, 400 mg (0.006 mM) of MIL-53(Fe) and 200 mg (0.0011 mol) of AgNO3 were added to 50 ml of deionized water and sonicated for 1 h. Afterwards, 20 ml of Na2HPO4 aqueous solution (0.0125 M) was added dropwise to the above solution. The total reaction mixture was stirred vigorously at room temperature for 6 h. The products were collected by filtration and were washed thoroughly with de-ionized water and dried overnight in a vacuum oven at 80 °C.2.4. Synthesis of the Ag/Ag3PO4/MIL-53(Fe) composite
The Ag/Ag3PO4/MIL-53(Fe) composite was prepared by a photo chemical reduction deposition method. In a typical synthetic procedure, the obtained Ag3PO4/MIL-53(Fe) composite was dispersed in 100 ml of double distilled water and the suspension was stirred for 20 min. The system was irradiated using a visible-light halogen linear lamp (500 W, 9500 Lumens) for 30 min to reduce Ag+ ions into Ag0 species on the surface of the Ag3PO4/MIL-53(Fe) composite. Finally, the product was collected by filtration, thoroughly washed with deionized water for several times, and dried in a vacuum oven at 80 °C overnight.2.5. Material characterization
The powder X-ray diffraction (PXRD) patterns were obtained using a Bruker D8 Advance X-ray diffractometer with CuKα (λ = 1.5406 Å) irradiation in the range of 10–70°. Scanning electron microscopy (SEM) images were recorded using a JEOL Model JSM – 6390LV (Japan Electron Optics Laboratory Co., Ltd, Japan). The transmission electron microscopy (TEM) measurement was conducted using a JEOL JEM 2100F microscope (Japan Electron Optics Laboratory Co., Ltd, Japan) working at an accelerating voltage of 200 kV. The UV-vis diffuse reflectance spectra (DRS) in the region of 300–800 nm were obtained using a Varian Cary 5000 spectrophotometer. The Fourier transform infrared (FTIR) spectra of the samples were recorded on a Thermo Nicolet Avatar 360 spectrometer using conventional KBr pellets.2.6. Evaluation of photocatalytic activity
The visible light photocatalytic activity of MIL-53(Fe), Ag3PO4, Ag3PO4/MIL-53(Fe), and Ag/Ag3PO4/MIL-53(Fe) composites was evaluated by studying the decolourization of an azo dye Ponceau BS aqueous solution under visible light irradiation. To know the origin of the reactive species involved in the photocatalytic oxidation process, phenol aqueous solution was also degraded under visible light irradiation over Ag/Ag3PO4/MIL-53(Fe). Typically, 0.3 g of the as-prepared photocatalyst was added to 300 ml of the dye aqueous solution and the suspension was stirred for 30 min under dark conditions to attain adsorption–desorption equilibrium between the catalyst and dye molecules. Finally, a 500 W tungsten halogen lamp was used as a visible light source to irradiate this dye aqueous solution containing the photocatalyst. 5 ml of the sample solution was sampled at every 10 min interval and centrifuged to remove the catalyst particles. A Shimadzu UV-vis 1601 was used to analyse the change in absorbance of the catalyst free dye solution samples as a function of irradiation time.3. Results and discussion
3.1. FTIR analysis
FTIR analysis was carried out to identify the functional group moieties of the as-prepared samples. Fig. 1 displays the FTIR spectra of MIL-53(Fe), Ag3PO4/MIL-53(Fe) and Ag/Ag3PO4/MIL-53(Fe).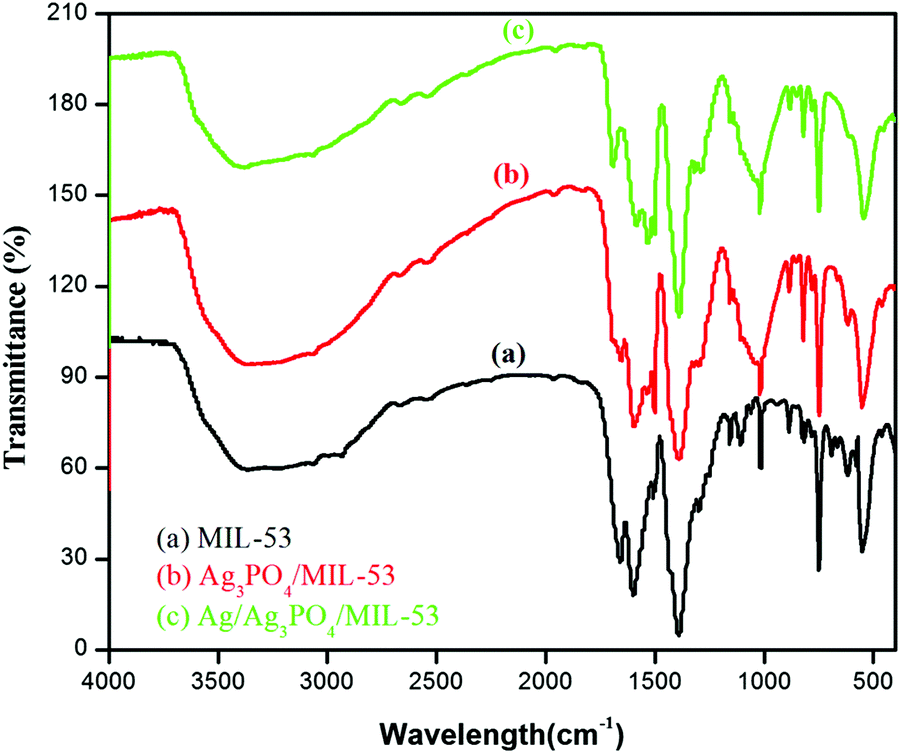 | ||
| Fig. 1 FTIR spectra of (a) MIL-53(Fe), (b) Ag3PO4/MIL-53(Fe) and (c) Ag/Ag3PO4/MIL-53(Fe). | ||
As shown in Fig. 1a, the observed peaks at 1598 cm−1 and 1398 cm−1 correspond to the asymmetric and symmetric vibrations of the carboxyl group, respectively.31 The peak at 750 cm−1 results from the C–H bending vibrations of the benzene ring.32,33 The formation of the metal–oxo bond between the Fe(III) and carboxylic group (Fe–O) is confirmed from the peak at 550 cm−1 which corresponds to the Fe–O bond. The two peaks at around 1000–1250 cm−1 in MIL-53(Fe) can be attributed to the C–N stretching of DMF molecules trapped in the pores of the MIL-53(Fe) framework.34,35 In Fig. 1b and c, the peaks corresponding to both MIL-53(Fe) and Ag3PO4 can be seen. The peaks at 830 cm−1 and 550 cm−1 are attributed to asymmetric stretching vibrations and bending vibrations of Ag3PO4 respectively.36 It is pertinent to mention here that in Fig. 1b and c, the peak at 550 cm−1 corresponding to the Fe–O bond of the MOF overlaps with the stretching vibrations of Ag3PO4. Furthermore, the peaks corresponding to the C–N stretching of DMF in pristine MIL-53(Fe) disappear in the composite which is attributed to the overlapping of these peaks with the broad band of P–O stretching.37
3.2. XRD analysis
In order to determine the crystallographic structure of the as-prepared samples, the materials were characterized by powder X-ray diffraction (PXRD). The XRD patterns of pristine MIL-53(Fe) and its corresponding composites are displayed in Fig. 2.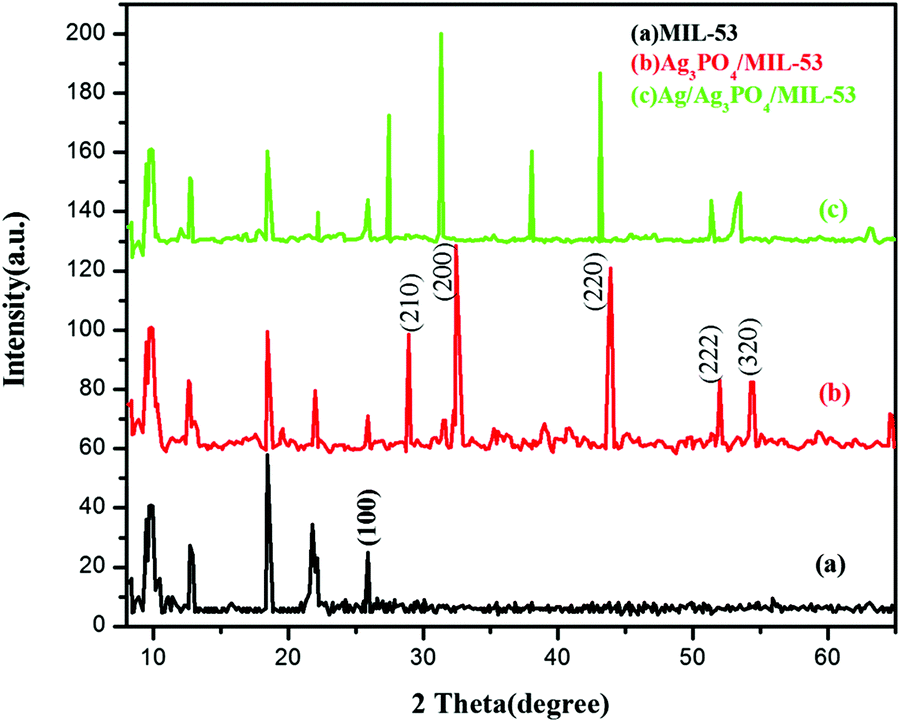 | ||
| Fig. 2 XRD patterns of (a) MIL-53(Fe), (b) Ag3PO4/MIL-53(Fe) and (c) Ag/Ag3PO4/MIL-53(Fe). | ||
As can be seen clearly from Fig. 2a, the diffraction peaks of pure MIL-53(Fe) at 2θ = 9.5°, 12.5°, 18.5° and 25.5° are in good agreement with the peaks already reported in the literature.38,39Fig. 2b and c displays the diffraction peaks of Ag3PO4/MIL-53(Fe) and Ag/Ag3PO4/MIL-53(Fe), respectively. The diffraction peaks at 28.0°, 32.5°, 37.8°, 44.3° and 64.2° in Fig. 2b can be indexed to the body-centred cubic structure of Ag3PO4. These diffraction peaks can be well indexed to the (210), (200), (220), (222) and (320) crystalline planes of Ag3PO4 suggesting the successful formation of the Ag3PO4/MIL-53(Fe) composite.40–42 In Fig. 2c, there is a slight shift in the peaks of Ag3PO4 towards left, which may be due to the incorporation of metallic silver in the framework resulting in the distortion of Ag3PO4. Furthermore, the diffraction peak at 2θ = 38.1° is assigned to metallic silver (Ag0) indicating that the Ag0 nanoparticles are generated on the surface of the catalyst.43 It is notable to mention here that the peak at 37.8° corresponding to Ag3PO4 has overlapped with the Ag0 peak at 38.1° giving rise to a very intense peak at 38.1°. Moreover, after incorporation of Ag3PO4, the main characteristic peaks corresponding to MIL-53(Fe) can be distinguished clearly in the composite catalyst indicating that MIL-53(Fe) remains intact during the synthesis of the composite catalyst.
The average crystallite size of the crystals was calculated using the Scherrer formula (eqn (1)).44
L = Kλ/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) cosθB cosθB | (1) |
| Sample | Average crystallite size (nm) | k app (min−1) | % Decolorization |
|---|---|---|---|
| MIL-53(Fe) | 28.2 | 0.01629 | 55 |
| Ag3PO4 | 0.0049 | 20 | |
| Ag3PO4/MIL-53(Fe) | 20.4 | 0.0249 | 71 |
| Ag/Ag3PO4/MIL-53(Fe) | 21.5 | 0.0460 | 94.8 |
3.3. Morphological characterization
FESEM and HRTEM analyses were used for morphological characterization of the samples. Fig. 3 shows the FESEM images of pure MIL-53(Fe) and Ag/Ag3PO4/MIL-53(Fe). | ||
| Fig. 3 FESEM image of (a and b) MIL-53(Fe) and (c and d) Ag/Ag3PO4/MIL-53(Fe). | ||
Fig. 3a and b displays the uniform polyhedron particle morphology of pure MIL-53(Fe) with an average particle diameter of 0.5 μm. As can be seen from Fig. 3a and b, there are only MIL-53(Fe) blocks confirming the phase purity of samples which is also confirmed from the TEM image of pure MIL-53(Fe) (Fig. 5a). This single phase purity is also in good agreement with the PXRD of MIL-53(Fe) (Fig. 2a). Fig. 3c and d shows the SEM images of Ag3PO4/MIL-53(Fe) and Ag/Ag3PO4/MIL-53(Fe) respectively. The uniform distribution of Ag3PO4 nanoparticles on the MOF surface facilitates better interaction at the interface. To further confirm the presence of Ag3PO4 in the composite, we performed EDX analysis of pristine MIL-53(Fe) and Ag/Ag3PO4/MIL-53(Fe). Fig. 4a displays the EDX spectrum of pure MIL-53(Fe) which shows the presence of C, O, and Fe, whereas the EDX spectrum of composite Ag/Ag3PO4/MIL-53(Fe) (Fig. 4b) clearly shows the presence of C, O, Fe, P, and Ag, thus proving the successful incorporation of Ag3PO4 onto MIL-53(Fe).
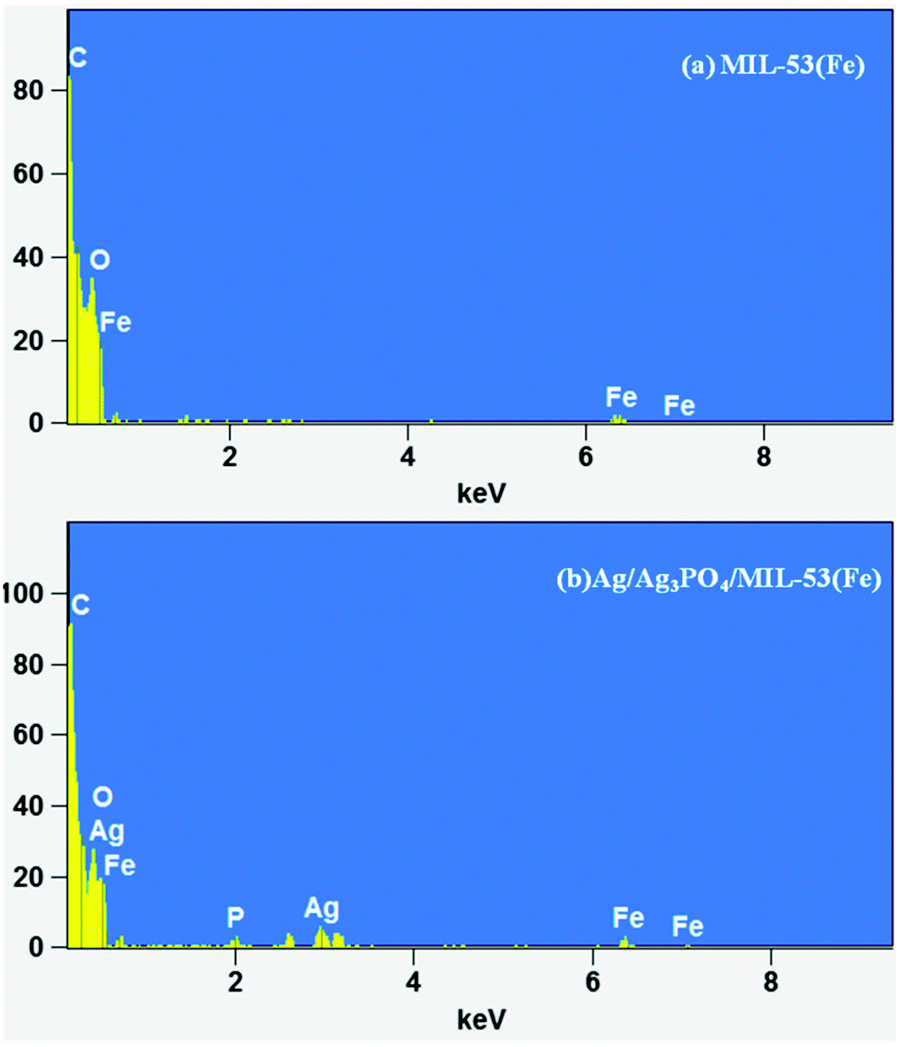 | ||
| Fig. 4 EDX spectrum of (a) MIL-53(Fe) and (b) Ag/Ag3PO4/MIL-53(Fe). | ||
In order to further ascertain the formation of a heterojunction, HRTEM analysis of pure MIL-53(Fe) and Ag/Ag3PO4/MIL-53(Fe) was carried out as shown in Fig. 5.
 | ||
| Fig. 5 (a) HRTEM image of MIL-53(Fe), (b) HRTEM image of Ag/Ag3PO4/MIL-53(Fe), (c) lattice spacing for Ag/Ag3PO4/MIL-53(Fe) and (d) SAED pattern of Ag/Ag3PO4/MIL-53(Fe). | ||
As can be seen clearly from Fig. 5a, MIL-53(Fe) exhibits uniform shape with size around 200 nm. Whereas from Fig. 5b, it is clear that the particles of Ag3PO4 are clearly deposited on MIL-53(Fe) confirming the formation of the heterostructure. The formation of the heterojunction between MIL-53(Fe) and Ag3PO4 is further confirmed from the lattice spacing between the adjacent fringes in the composite catalyst (Fig. 5c). The spacing of lattice fringes was found to be 0.48 nm and 0.16 nm which are consistent with the crystal planes of MIL-53(Fe) and Ag3PO4 respectively. The above results clearly indicate the formation of the heterojunction. Fig. 5d displays the selected area electron diffraction pattern (SAED) of Ag/Ag3PO4/MIL-53(Fe). It can be seen clearly from Fig. 5d that there are clear bright diffraction spots and rings confirming the crystalline nature of the material. From Fig. 5d, two main diffraction rings can be indexed as the characteristic rings of the (100) plane of MIL-53(Fe) which is consistent with the results of the XRD pattern (Fig. 2a).
3.4. Thermal studies
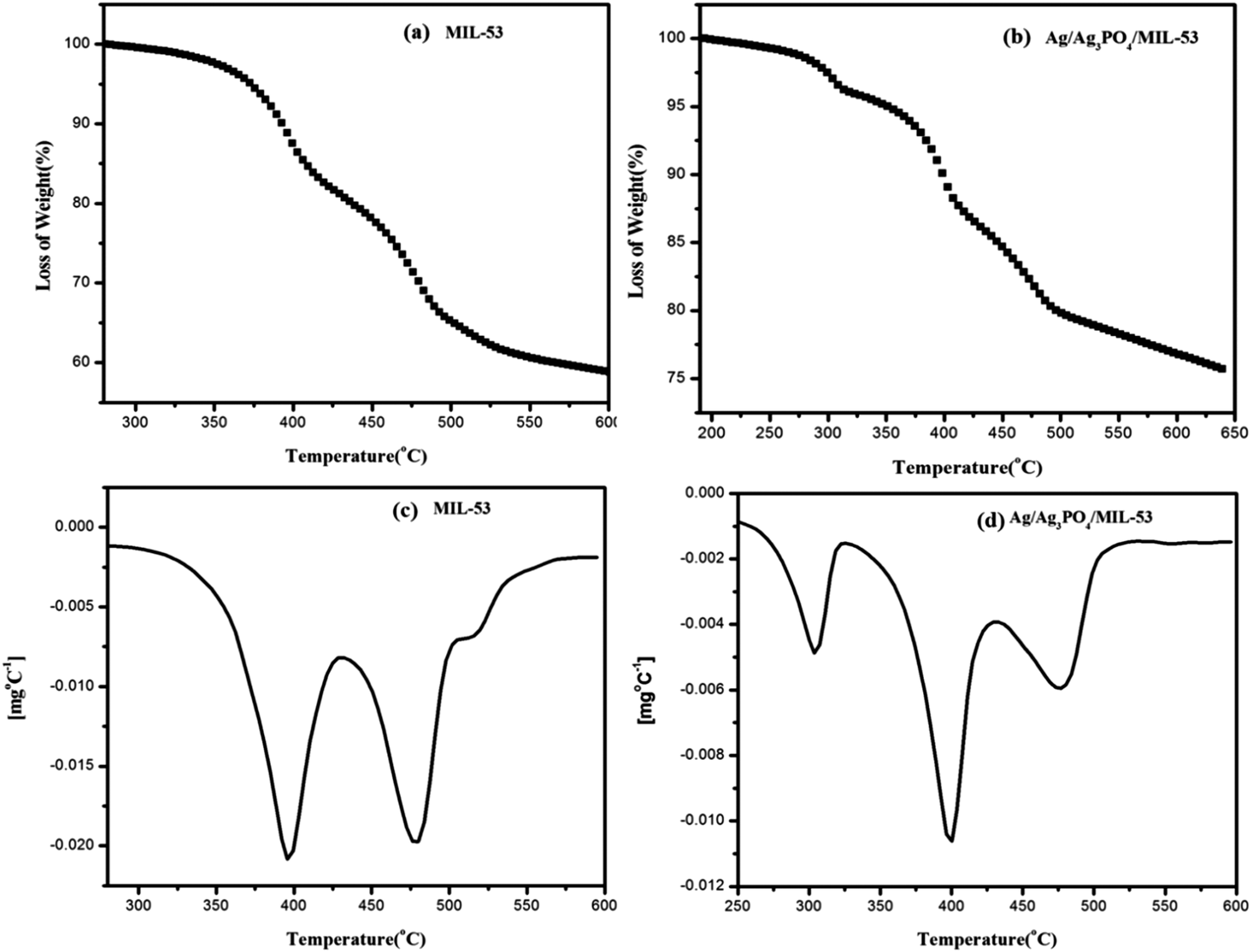 | ||
| Fig. 6 TGA curves of (a) MIL-53(Fe), (b) Ag/Ag3PO4/MIL-53(Fe) and DTG curves of (c) MIL-53(Fe) and (d) Ag/Ag3PO4/MIL-53(Fe), respectively. | ||
Fig. 6 depicts the multistep decomposition of both the samples owing to the preferential loss of the moieties. For pristine MIL-53(Fe) (Fig. 6a), it is clear that the metal–organic-framework is stable up to 350 °C which is consistent with the already reported literature.45 The TGA curve in the temperature range of 350–400 °C involves a sharp decrease in weight (20%), which is preferably due to the loss of free DMF molecules inside the pores of MIL-53(Fe). Furthermore, the weight loss between 400 and 500 °C is due to the framework decomposition involving the elimination of organic links within the framework. This slow decomposition rate from 350 °C onwards is presumably due to the formation of Fe2O3 molecules45 hence acting as a barrier for heat transfer to the metal–organic-framework. In the case of Ag/Ag3PO4/MIL-53(Fe) (Fig. 6b), the weight loss between 200 and 300 °C is attributed to the decomposition of silver nanoparticles present on the surface of the composite material. It can be seen clearly from Fig. 6a that for pristine MIL-53(Fe), 40% of decomposition occurs in the window of 280–600 °C, whereas in the case of Ag/Ag3PO4/MIL-53(Fe) (Fig. 6b) only 22% decomposition occurs in the same temperature range indicating the high thermal stability of the material. This increase in thermal stability of MIL-53(Fe) by functionalization with Ag/Ag3PO4 is attributed to the strengthening of the organic linker attached to iron due to the interaction of silver nanoparticles with the framework.
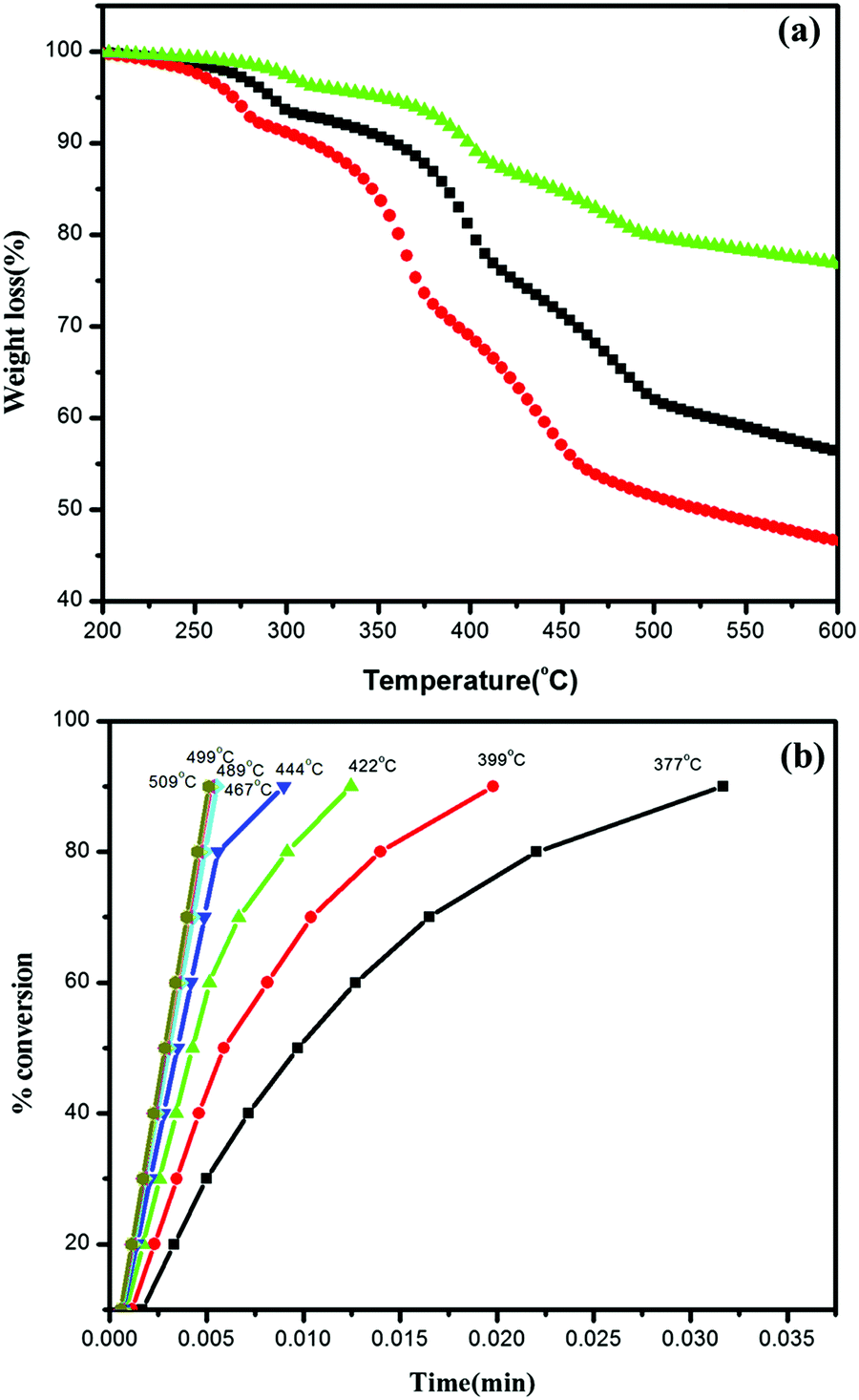 | ||
| Fig. 7 (a) TGA curves of Ag/Ag3PO4/MIL-53(Fe) at three different heating rates, 10 °C, 15 °C, and 20 °C and (b) degree of decomposition at different temperature steps versus time. | ||
It can be seen clearly from Fig. 7a that decomposition is a multistep with the maximum rate of framework decomposition in the range of 377–509 °C involving the elimination of the organic linker. The kinetic parameters of the framework decomposition step were calculated using Star e software with activation energy Ea = 80 kJ mol−1 and n = 1. Furthermore, the degree of conversion was also calculated within the same temperature window as illustrated in Fig. 7b, with the rate constants simulated in Table 2, at each temperature step in the window indicating the first order kinetics of decomposition.
| Temperature (°C) | TMR × 10−03 | ln(k) |
|---|---|---|
| 377 | 13.6 | 0.2 |
| 399 | 8.4 | 0.69 |
| 422 | 5.2 | 1.16 |
| 444 | 3.4 | 1.58 |
| 467 | 2.3 | 2.0 |
| 489 | 1.5 | 2.38 |
| 500 | 1.3 | 2.54 |
| 509 | 1.1 | 2.70 |
3.5. Optical properties
UV-vis diffuse reflectance spectroscopy (DRS) was used to study the optical properties of materials. The DRS of the samples and the corresponding band gaps are shown in Fig. 8 and 9.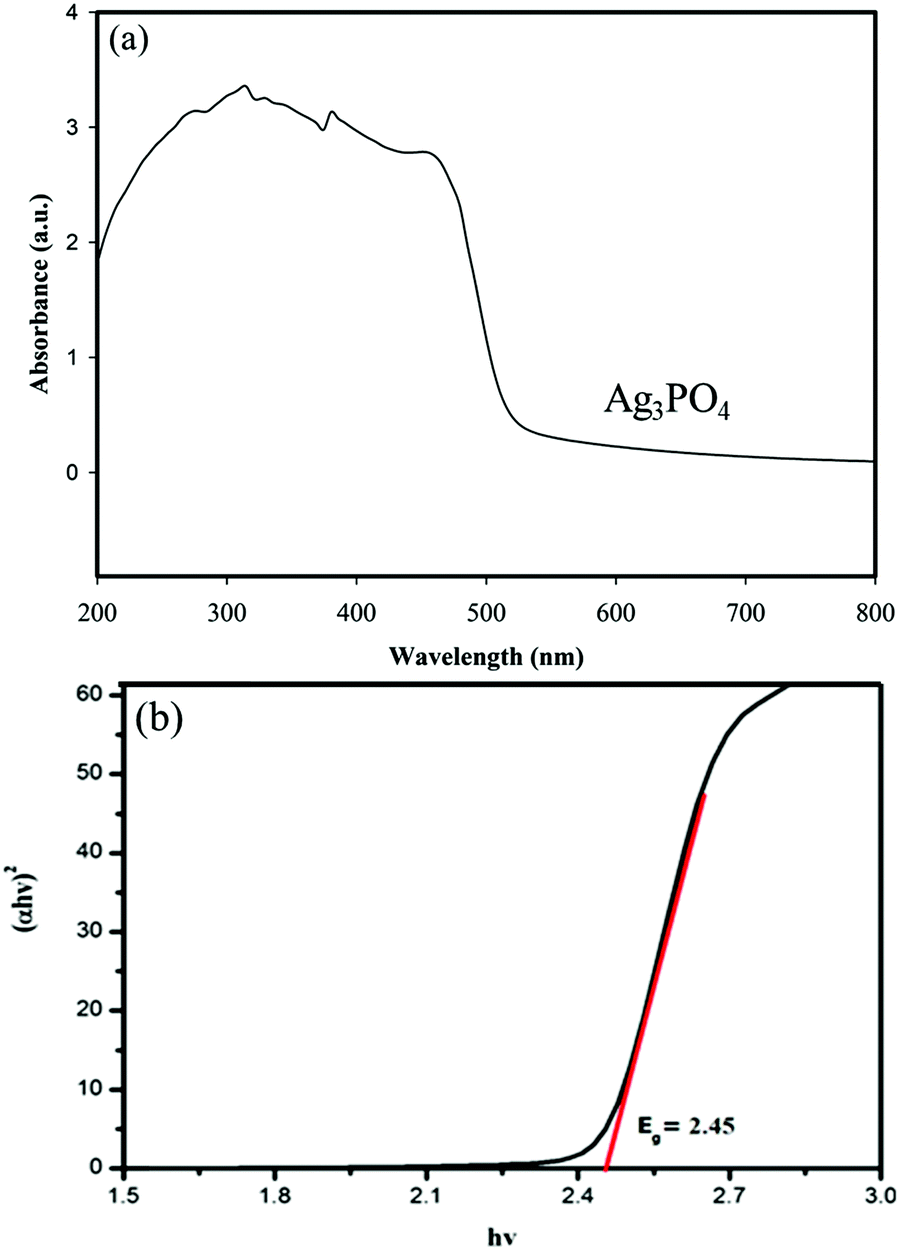 | ||
| Fig. 8 (a) UV-vis diffuse reflectance spectra of Ag3PO4 and (b) plot of (αhν)2vs. photon energy (hν) for the band gap energy of Ag3PO4. | ||
 | ||
| Fig. 9 (a) UV-vis diffuse reflectance spectra of MIL-53(Fe) and Ag/Ag3PO4/MIL-53(Fe), (b) plots of (αhv)2vs. photon energy (hν) for the band gap energy of MIL-53(Fe) and Ag/Ag3PO4/MIL-53(Fe). | ||
Fig. 8a shows that the absorption edge of Ag3PO4 is around 560 nm.
The optical band gap of the samples was calculated from the following formula reported by Butler:46
| αhν = A(hν − Eg)n/2 | (2) |
It can be seen clearly from Fig. 9a that the absorption edge of pure MIL-53(Fe) and Ag/Ag3PO4/MIL-53(Fe) is around 650–70 nm. Moreover, it can be seen clearly from Fig. 9a that Ag/Ag3PO4/MIL-53(Fe) has better light absorption which is attributed to the functionalization of MIL-53(Fe) by Ag3PO4. The band gap of pure MIL-53(Fe) and Ag/Ag3PO4/MIL-53(Fe) was calculated to be 2.0 and 1.8 eV, respectively (Fig. 9b), indicating the better light absorption of Ag/Ag3PO4/MIL-53(Fe).
3.6. Photocatalytic activity
Fig. 10 shows the experimental results of the photocatalytic degradation of Ponceau BS and phenol using various synthesized catalytic systems.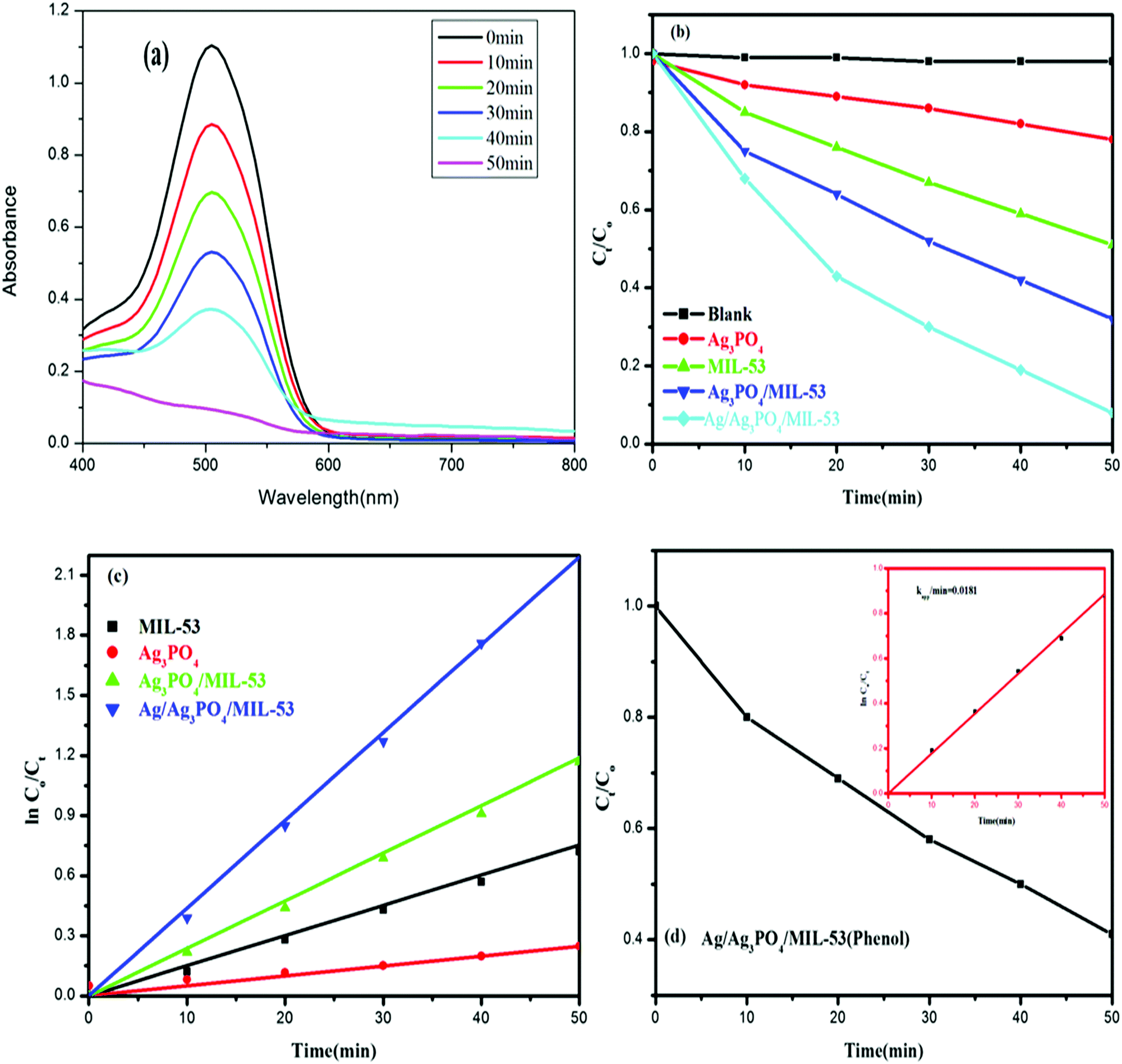 | ||
| Fig. 10 (a) UV-vis spectral changes of the degradation of PBS by Ag/Ag3PO4/MIL-53(Fe), (b) the degradation efficiency (Ct/C0) of PBS in the presence of MIL-53(Fe), Ag3PO4, Ag3PO4/MIL-53(Fe) and Ag/Ag3PO4/MIL-53(Fe), (c) PBS decolourization curves of ln(C0/Ct) versus irradiation time for different catalysts and (d) the degradation efficiency (Ct/C0) of phenol in the presence of Ag/Ag3PO4/MIL-53(Fe); inset shows ln(C0/Ct) versus irradiation time. | ||
The observed spectral changes of PBS at its λmax over the Ag/Ag3PO4/MIL-53(Fe) catalyst (Fig. 10a) depict a significant decrease in its characteristic absorption band. Fig. 10b displays the photocatalytic performance of pristine MIL-53(Fe), Ag3PO4, Ag3PO4/MIL-53(Fe) and Ag/Ag3PO4/MIL-53(Fe) over degradation of PBS (Ct/C0) under visible light irradiation. Control experiments in the absence of catalyst and light irradiation were also carried out. The self-degradation of PBS in the absence of catalyst under visible light irradiation was found to be insignificant. Besides, dark adsorption was monitored for 30 min to attain adsorption–desorption equilibrium. It is obvious from Fig. 10b that among the as-synthesized photocatalysts, Ag/Ag3PO4/MIL-53(Fe) showed the best photocatalytic efficiency and degraded 94.8% PBS in just 50 min. On the other hand, pure MIL-53(Fe) and Ag3PO4/MIL-53(Fe) degrade 55% and 71% of PBS, respectively. Phenol, a colourless molecule, was selected as a probe to ensure that the reactive species are generated from the catalyst and not from the photosensitization process. H2O2 was added to the reaction mixture as an external electron acceptor species. It can be seen from Fig. 10d that the concentration of phenol (Ct/C0) in the presence of Ag/Ag3PO4/MIL-53(Fe) decreases with increasing irradiation time suggesting that the reactive species originate from the photocatalyst and hence ruling out the possibility of photosensitization of dye PBS.
Quantitatively, different degradation rates were simulated on the basis of the Langmuir–Hinshelwood kinetic model, assuming that all the reactions follow the pseudo-first order kinetics model given by eqn (3)
| ln(C0/Ct) = kt | (3) |
3.7. Proposed mechanism
To investigate the photocatalytic mechanism involved in the degradation of Ponceau BS over Ag/Ag3PO4/MIL-53(Fe), trapping experiments were carried out to investigate the possible reactive species involved in the photocatalytic oxidation process. Benzoquinone (BQ), ethylenediamminetetraacetic acid (EDTA) and isopropyl alcohol (IPA) were introduced into the reaction system to quench superoxide (˙−O2), hole (h+) and hydroxyl (˙OH), respectively47 and the results are displayed in Fig. 11.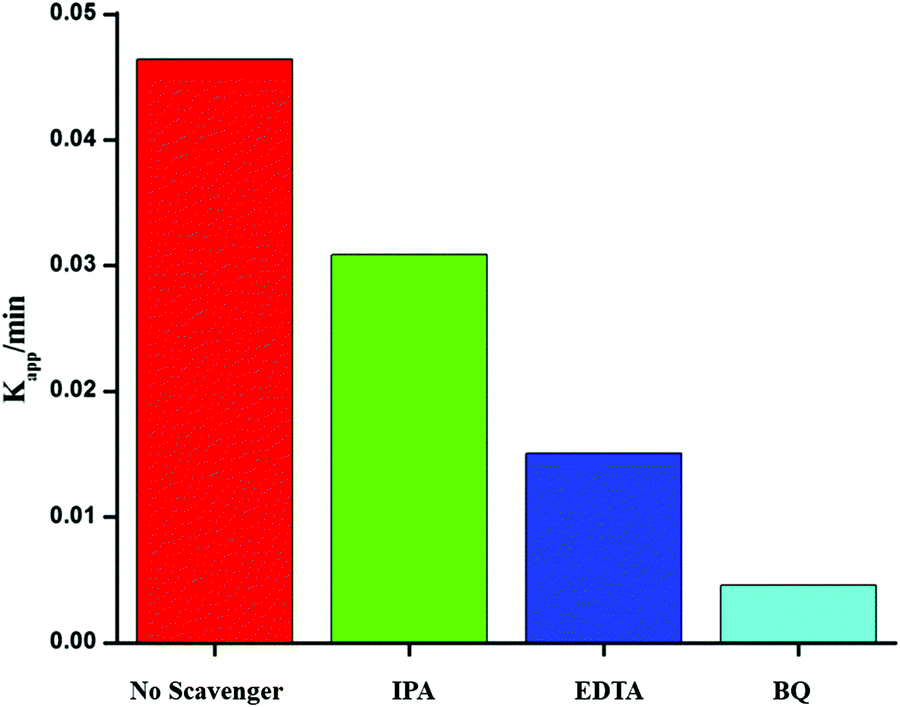 | ||
| Fig. 11 Effects of different scavengers on the degradation of PBS over the Ag/Ag3PO4/MIL-53(Fe) catalyst; BQ = benzoquinone, IA = isopropyl alcohol, and EDTA = ethylenediamminetetra acetic acid. | ||
From Fig. 11, it is observed that the addition of BQ and EDTA decreases the rate of degradation significantly compared to without addition of a scavenger under similar conditions whereas IPA had a little effect on the rate of degradation of PBS. From these results, it is clear that superoxide and hole are primary reactive species involved in the photocatalytic degradation of PBS under visible light.
The valence band (VB) edge potential of the photocatalysts was calculated using the empirical equation.48
| EVB = X − Ee + 0.5Eg | (4) |
| ECB = EVB − Eg | (5) |
 | ||
| Fig. 12 Schematic diagram showing the band structure and Z-scheme separation of photoinduced electron–hole pairs at the interface of the Ag/Ag3PO4/MIL-53(Fe) catalyst under visible light irradiation. | ||
Upon visible light irradiation, both Ag3PO4 and MIL-53(Fe) generate electron–hole pairs in the conduction and valence band. Due to excellent conductivity, the photogenerated electrons are quickly transferred from the conduction band of Ag3PO4 to the silver nanoparticles and simultaneously the holes from the VB of MIL-53(Fe) move to the VB of Ag NPs thereby combining with electrons. Meanwhile, the photogenerated electrons in the CB of MIL-53(Fe) are continuously trapped by O2 to form ˙−O2 which further oxidizes the organic contaminants. Secondly, the holes present in the VB of Ag3PO4 react with H2O molecules to form ˙OH which directly reacts with the organic pollutant for further degradation. Therefore, enhancement in the photocatalytic activity of Ag/Ag3PO4/MIL-53(Fe) for degradation of PBS is mainly due to the internal charge transmission from MIL-53(Fe) and Ag3PO4 to the Ag NPs clusters via the Z-scheme pathway.
3.8. Stability of the catalyst
For practical applications, the stability of the photocatalyst is the main factor. Besides, silver-based photocatalysts are very much unstable. Keeping in view the instability of the ‘Ag’-based photocatalyst, Ag/Ag3PO4/MIL-53(Fe) was regenerated by centrifugation after the degradation reaction and was washed with water three times to eliminate contaminants on the surface. The catalyst was then reused for four cyclic runs under the same conditions. The results are displayed in Fig. 13, which clearly indicate that PBS degradation shows only a slight decline after four cycling experiments and PBS degradation is maintained at 91.1% confirming that Ag/Ag3PO4/MIL-53(Fe) is a highly stable photocatalyst.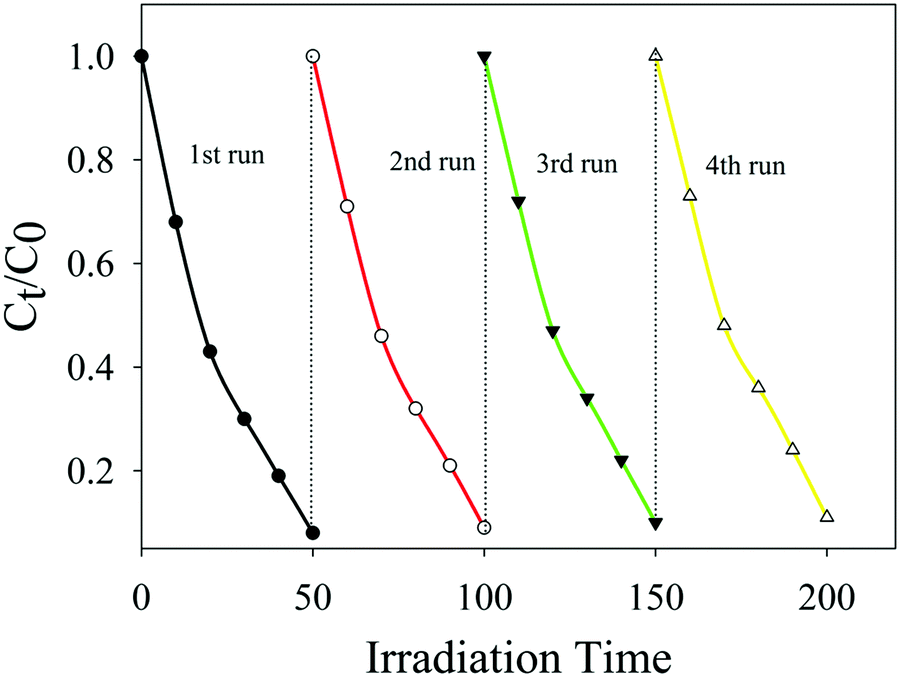 | ||
| Fig. 13 The repeated experiments of photocatalytic degradation of PBS over the Ag/Ag3PO4/MIL-53(Fe) catalyst. | ||
The stability and better efficiency of the composite photocatalyst are attributed to the positive synergistic effects of Ag3PO4 and MIL-53(Fe) on the effective separation of photo-generated charge carriers. Due to these factors, the electrons and holes effectively migrate from the CB and VB of the catalyst to the surface, thereby reducing the chances of erosion of the catalyst.
4. Conclusion
In summary, a facile microwave method was used for the synthesis of novel double-visible-light active components, MIL-53(Fe)-based Ag/Ag3PO4/MIL-53(Fe) photocatalysts. The Ag/Ag3PO4/MIL-53(Fe) nanocomposite photocatalysts display superior photocatalytic performance for the degradation of organic contaminants compared to pristine MIL-53(Fe) and Ag3PO4/MIL-53(Fe) under visible light irradiation. On the basis of the trapping experiment results and the calculation of band potential values, it was proposed that the formation of Ag nanoparticles on the surface of the catalyst improves the photocatalytic ability substantially via the Z-scheme internal charge transmission mechanism. These Ag nanoparticles act as a recombination centre for the electrons of Ag3PO4 and holes of MIL-53(Fe) leaving electrons of MIL-53(Fe) on CB and holes of Ag3PO4 on VB. The Kapp values obtained clearly suggest that the rate of degradation of PBS over Ag/Ag3PO4-MIL-53(Fe) (0.001 min−1) is 5 and 7 times higher than Ag3PO4-MIL-53(Fe) and pristine MIL-53(Fe), respectively. Moreover, the catalyst was found to be stable even after 4 recycling runs. The present contribution provides an insight into the role of metal nanoparticles in improving the photocatalytic ability of metal–organic-frameworks, a field that has remained unexplored.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are highly thankful to the Department of chemistry, NIT, Srinagar, and the Central Research Facility, NIT, Srinagar, for providing instrumentation facility. SAIF Cochin, Kerala, is also acknowledged for providing instrumentation facility for sample analysis. Feroz Ahmad Sofi is also thankful to MHRD, Delhi, India, for providing financial assistance.References
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- J.-W. Liu, L.-F. Chen, H. Cui, J.-Y. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- A. Dhakshinamoorthy, A. M. Asiri and H. Garćıa, Angew. Chem., Int. Ed., 2016, 55, 2–35 CrossRef PubMed.
- M. D. Allendorf, C. A. Bauer, R. K. Bhaktaa and R. J. T. Houka, Chem. Soc. Rev., 2009, 38, 1330–1352 RSC.
- J. Zhuang, C.-H. Kuo, L.-Y. Chou, D.-Y. Liu, E. Weerapana and C.-K. Tsung, ACS Nano, 2014, 8, 2812–2819 CrossRef CAS PubMed.
- M. Alvaro, E. Carbonell, B. Ferrer, F. X. Llabrés i Xamena and H. Garcia, Chem. – Eur. J., 2007, 13, 5106–5112 CrossRef CAS PubMed.
- C. G. Silva, A. Corma and H. Garcia, J. Mater. Chem., 2010, 20, 3141–3156 RSC.
- R. Li, J. Hu, M. Deng, H. Wang, X. Wang, Y. Hu, H.-L. Jiang, J. Jiang, Q. Zhang, Y. Xie and Y. Xiong, Adv. Mater., 2014, 26, 4783–4788 CrossRef CAS PubMed.
- M. C. Das, H. Xu, Z. Wang, G. Srinivas, W. Zhou, Y.-F. Yue, V. N. Nesterov, G. Qian and B. Chen, Chem. Commun., 2011, 47, 11715–11717 RSC.
- M. Fuentes-Cabrera, D. M. Nicholson, B. G. Sumpter and M. Widom, J. Chem. Phys., 2005, 123, 124713 CrossRef PubMed.
- S.-L. Li and Q. Xu, Energy Environ. Sci., 2013, 6, 1656–1683 CAS.
- K. Manna, T. Zhang, F.-X. Greene and W.-B. Lin, J. Am. Chem. Soc., 2015, 137, 2665–2673 CrossRef CAS PubMed.
- C.-C. Wang, J.-R. Li, X.-L. Lv, Y.-Q. Zhang and G.-S. Guo, Energy Environ. Sci., 2014, 7, 2831–2867 CAS.
- A. Nasalevich, M. G. Goesten, T. J. Savenije, F. Kapteijna and J. Gascon, Chem. Commun., 2013, 49, 10575–10577 RSC.
- D.-S. Chen, H.-Z. Xing, C.-G. Wang and Z.-M. Su, J. Mater. Chem. A, 2016, 4, 2657–2662 CAS.
- W.-Y. Gao, M. Chrzanowski and S.-Q. Ma, Chem. Soc. Rev., 2014, 43, 5841–5866 RSC.
- S. Huh, S.-J. Kim and Y. Kim, CrystEngComm, 2016, 18, 345–368 RSC.
- X.-Y. Li, Y.-H. Pi, Q.-B. Xia, Z. Li and J. Xiao, Appl. Catal., B, 2016, 191, 192–201 CrossRef CAS.
- C. Wang, K. E. deKrafft and W.-B. Lin, J. Am. Chem. Soc., 2012, 134, 7211–7214 CrossRef CAS PubMed.
- Z.-W. Yang, X.-Q. Xu, X.-X. Liang, C. Lei, Y.-L. Wei, P.-Q. He, B.-L. Lv, H.-C. Ma and Z.-Q. Lei, Appl. Catal., B, 2016, 198, 112–123 CrossRef CAS.
- Z. Sha, J. Sun, H. S. O. Chan, S. Jaenicke and J. Wu, RSC Adv., 2012, 1–3 Search PubMed.
- E. Haque, J. W. Jun and S. H. Jhung, J. Hazard. Mater., 2011, 185, 507–511 CrossRef CAS PubMed.
- M. Anbia, V. Hoseini and S. Sheykhi, J. Ind. Eng. Chem., 2012, 18, 1149–1152 CrossRef CAS.
- Y. Bi, S. Ouyang, J. Cao and J. Ye, Phys. Chem. Chem. Phys., 2011, 13, 10071–10075 RSC.
- G. F. Huang, Z. L. Ma, W. Q. Huang, Y. Tian, C. Jiao, Z. M. Yang and A. Pan, J. Nanomater., 2013, 1 Search PubMed.
- C. Hu, T. Peng, X. Hu, Y. Nie, X. Zhou, J. Qu and H. He, J. Am. Chem. Soc., 2009, 132, 857–862 CrossRef PubMed.
- C. Hu, T. Peng, X. Hu, Y. Nie, X. Zhou, J. Qu and H. He, J. Am. Chem. Soc., 2009, 132, 857–862 CrossRef PubMed.
- W. Wang, B. Cheng, J. Yu, G. Liu and W. Fan, Chem. – Asian J., 2012, 7, 1902–1908 CrossRef CAS PubMed.
- Z. C. Wang, J. H. Liu and W. Chen, Dalton Trans., 2012, 41, 4866–4870 RSC.
- C. Zhang, L. Ai and J. Jiang, Ind. Eng. Chem. Res., 2015, 54, 153–163 CrossRef CAS.
- P. Horcajada, C. Serre, G. Maurin, N. A. Ramsahye, F. Balas, R. M. Vallet and G. Férey, J. Am. Chem. Soc., 2008, 130, 6774–6780 CrossRef CAS PubMed.
- A. Banerjee, R. Gokhale, S. Bhatnagar, J. Jog, M. Bhardwaj, B. Lefez and S. Ogale, J. Mater. Chem., 2012, 22, 19694–19699 RSC.
- K. Zarshenas, A. Raisi and A. Aroujalian, RSC Adv., 2015, 5, 19760–19772 RSC.
- G. Lian, X. Zhang, M. Tan, S. Zhang, D. Cui and Q. Wang, J. Mater. Chem., 2011, 21, 9201–9207 RSC.
- C. Gong, D. Chen, X. Jiao and Q. Wang, J. Mater. Chem., 2002, 12, 1844–1847 RSC.
- S. S. Das, N. P. Singh, V. Srivastava and P. K. Srivastava, Indian J. Eng. Mater. Sci., 2008, 15, 256–264 CAS.
- L. Ai, L. Li, C. Zhang, J. Fu and J. Jiang, Chem. – Eur. J., 2013, 19, 15105–15108 CrossRef CAS PubMed.
- R. Liang, F. Jing, L. Shen, N. Qin and L. Wu, J. Hazard. Mater., 2015, 287, 364–372 CrossRef CAS PubMed.
- Y. Liu, L. Fang, H. Lu, L. Liu, H. Wang and C. Hu, Catal. Commun., 2012, 17, 200–204 CrossRef CAS.
- J. Tang, W. Gong, T. Cai, T. Xie, C. Deng, Z. Peng and Q. Deng, RSC Adv., 2013, 3, 2543–2547 RSC.
- Y. Gu, X. Gu, Y. Zhao and Y. Qiang, J. Mater. Sci.: Mater. Electron., 2015, 26, 5237–5242 CrossRef CAS.
- J. Cao, B. Luo, H. Lin, B. Xu and S. Chen, J. Hazard. Mater., 2012, 217, 107–115 CrossRef PubMed.
- J. Cao, B. Xu, H. Lin, B. Luo and S. Chen, Chem. Eng. J., 2012, 185, 91–99 CrossRef.
- R. Panda, S. Rahut and J. K. Basu, RSC Adv., 2016, 6, 80981 RSC.
- M. A. Butler, J. Appl. Phys., 1977, 48, 1914–1920 CrossRef CAS.
- G. Li, K. H. Wong, X. Zhang, C. Hu, C. Y. Jimmy, R. C. Y. Chan and P. K. Wong, Chemosphere, 2009, 76, 1185–1191 CrossRef CAS PubMed.
- X. Zhang, L. Zhang, T. Xie and D. Wang, J. Phys. Chem. C, 2009, 113, 7371–7378 CAS.
- X. Wang, S. Li, Y. Ma, H. Yu and J. Yu, J. Phys. Chem. C, 2011, 115, 14648–14655 CAS.
- A. J. Bard, R. Parsons and J. Jordan, Standard potentials in aqueous solutions, CRC Press, New York, 1985 Search PubMed.
| This journal is © the Partner Organisations 2018 |