
Aggregation-induced emission-mediated spectral downconversion in luminescent solar concentrators†
Bolong
Zhang
 a,
James L.
Banal
b,
David J.
Jones
a,
Ben Zhong
Tang
c,
Kenneth P.
Ghiggino
a and
Wallace W. H.
Wong
*a
a,
James L.
Banal
b,
David J.
Jones
a,
Ben Zhong
Tang
c,
Kenneth P.
Ghiggino
a and
Wallace W. H.
Wong
*a
aSchool of Chemistry, Bio21 Institute, The University of Melbourne, Parkville, Victoria 3010, Australia. E-mail: wwhwong@unimelb.edu.au
bDepartment of Biological Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA
cHong Kong Branch of Chinese National Engineering Research Centre for Tissue Restoration and Reconstruction, Department of Chemistry, The Hong Kong University of Science & Technology (HKUST), Clear Water Bay, Kowloon, Hong Kong
First published on 29th January 2018
Abstract
The light-harvesting efficiency of luminescent solar concentrators (LSCs) is encumbered by reabsorption of emission and concentration quenching. Energy transfer from a high-concentration donor to a low-concentration energy trap can reduce reabsorption losses while maintaining efficient light collection. Emissive aggregates enable this approach by reducing the impact of concentration quenching, which is detrimental to the entire energy transfer process. Here we describe a LSC that utilizes emissive aggregates as energy-transfer pairs for downconversion. We characterize the photophysics of a benzothiadiazole-based emissive aggregate, PITBT-TPE, that complements a highly emissive donor, DPATPAN, and functions as a highly emissive energy-transfer acceptor even at high concentrations in excess of 180 mM in the PMMA matrix. Monte Carlo simulations of LSCs that leverage these emissive aggregates as energy-transfer pairs predicted notable optical efficiencies at large concentrator dimensions. We demonstrate for the first time a LSC that utilizes donor and acceptor AIE chromophores to reduce reabsorption.
Widespread utility of solar technologies requires lowering the cost of highly-efficient photovoltaics devices while also ensuring compatibility and integration with urban infrastructures, such as buildings, houses, or noise-barriers.1–4 Luminescent solar concentrators (LSCs) are large-area and planar spectral downconverting devices that function as a light-harvesting accessory to small-area photovoltaic cells.5 A typical LSC consists of a planar glass or plastic waveguide with chromophores dispersed on the surface of a waveguide or embedded within a high refractive index matrix for waveguiding (Fig. 1a). The chromophores absorb incident photons and subsequent emitted photons can be trapped in the waveguide by total internal reflection. The emitted photons are concentrated to the edge of the waveguide where a photovoltaic cell is attached.5,6 As the emitted photons traverse through the waveguide, the probability of reabsorption by other chromophores increases due to multiple total internal reflection events. These reabsorbed photons can be lost by non-radiative decay, if the photoluminescence quantum yield (ϕPL) of the chromophore is less than unity, and by emission into the escape cone of the waveguide. Chromophores that have innately large Stokes shifts have been proposed to circumvent the reabsorption.7,8 However, further increasing the Stokes shift by introducing electron donating/withdrawing substituents to the chromophore often leads to a compromise with quantum yields.8 A large Stokes shift can also be achieved using a Förster resonance energy transfer (FRET) donor–acceptor pair. An emissive donor of significantly high concentration within the LSC matrix absorbs light and transfers its energy non-radiatively via Förster mechanism to an acceptor that has a comparatively lower concentration in the film.9–12 The acceptor then emits at longer wavelengths compared to the incident light. By optimizing the concentration and ratio of donor and acceptor chromophores in the LSC, reabsorption from the acceptor can be mitigated while maintaining efficient light-harvesting.9–11,13 Using this energy migration and trapping approach for LSCs provides broad tunability in the emission wavelengths and absorption bandwidth of LSCs.
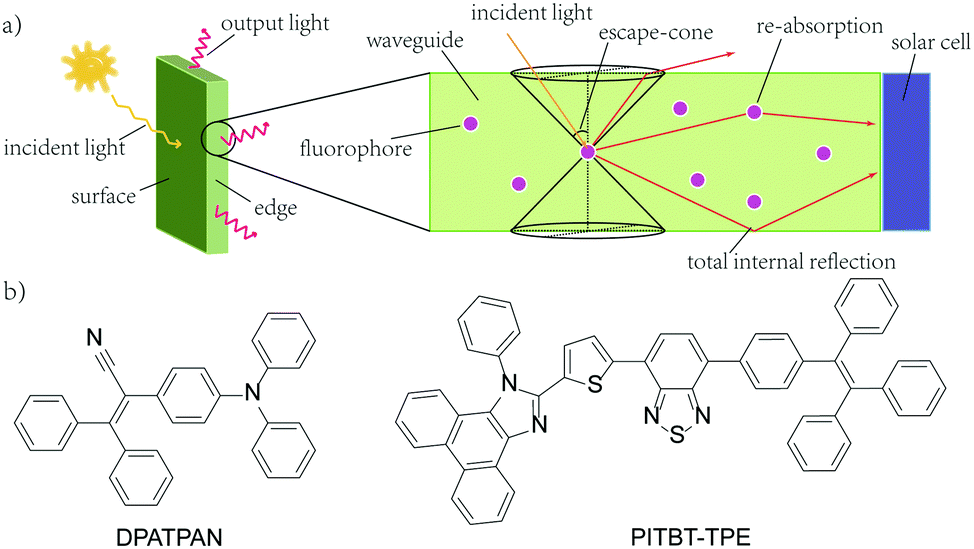 | ||
| Fig. 1 (a) A typical structure of a LSC with attached solar cell (b) the chemical structures of DPATPAN and PITBT-TPE. | ||
Our group recently reported the use of FRET donor chromophores that showed high ϕPL at high concentration in poly(methyl methacrylate) (PMMA).9 Previously, we have identified a chromophore, DPATPAN that has aggregation-induced emission (AIE) characteristics14 and was used as the FRET donor with a laser dye, DCJTB, as the FRET acceptor (structure of DPATPAN shown in Fig. 1b).9 While this FRET system showed enhanced LSC performance relative to DCJTB alone, DCJTB underwent fast photobleaching in ambient experimental conditions, in addition to concentration quenching, which limits its long-term utility for LSCs. We identified benzothiadiazole-based PITBT-TPE, a recently synthesized AIE chromophore,15 as a potential acceptor candidate to replace DCJTB. Apart from being an appropriate FRET acceptor for DPATPAN, we observed that PITBT-TPE is still highly emissive even at very high concentrations in PMMA. Here, we investigated the photophysical properties and LSC performance of the DPATPAN/PITBT-TPE donor–acceptor pair. A high quantum yield was observed for PITBT-TPE and its blends with DPATPAN even at very high concentrations. Monte Carlo ray tracing simulations were used to predict the possible performance of a LSC that uses the DPATPAN/PITBT-TPE energy-transfer pair. We demonstrate for the first time LSC devices that utilize a FRET pair with both chromophores showing AIE characteristics.
All thin-film samples in this work were PMMA films containing chromophores which were deposited on glass slides by spin-coating from chloroform solutions (see ESI† for sample preparation details). The absorption spectrum of PITBT-TPE overlaps well with the emission spectrum of DPATPAN (Fig. 2a). In addition, the emission onset for PITBT-TPE in PMMA is significantly shifted to longer wavelengths relative to the absorption tail of DPATPAN, similar to DCJTB. This spectral separation between the donor absorbance and acceptor emission spectra is key to avoid reabsorption of acceptor emission by the donor, particularly for the energy migration and trapping approach wherein the tail optical density of the donor can be significant to maintain efficient light-harvesting. Significant tail absorption, as demonstrated by us16 and others,17 can lead to considerable reabsorption losses in LSCs.
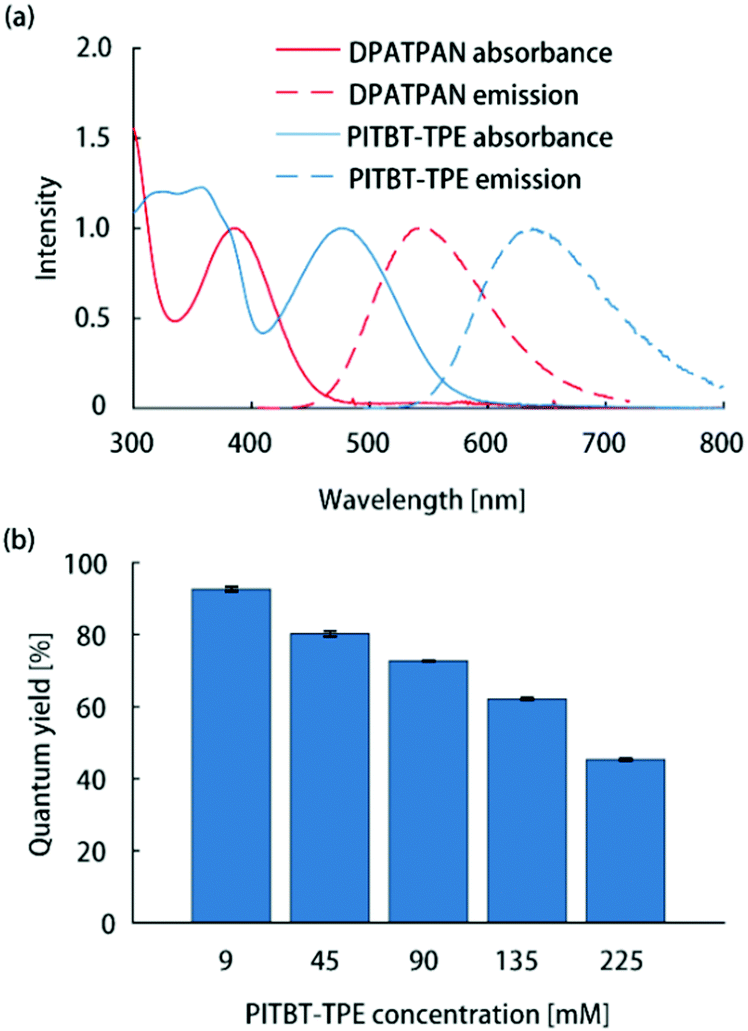 | ||
| Fig. 2 (a) Normalized absorption and emission spectra of DPATPAN (250 mM in PMMA) and PITBT-TPE (200 mM in PMMA). (b) Emission quantum yield of PITBT-TPE at different concentrations in PMMA. | ||
The ϕPL measurements of chromophore thin-films were carried out using the integrating sphere method (see ESI† for details).13 The quantum yield of PITBT-TPE in a PMMA thin-film matrix decreased as a function of concentration, from 93% at 9 mM to 45% at 225 mM (Fig. 2b). This suggested that PITBT-TPE, despite being an AIE chromophore, was susceptible to concentration quenching in a PMMA matrix. In contrast, DPATPAN, also an AIE chromophore, showed much higher concentration quenching tolerance compared to PITBT-TPE. The ϕPL of DPATPAN was ∼98% even at 250 mM in PMMA.
Using a constant DPATPAN concentration of 250 mM in PMMA, the ϕPL of films containing different DPATPAN and PITBT-TPE ratios was measured (Fig. 3a). The emission spectra of the blends systematically shifted to longer wavelengths with increasing amounts PITBT-TPE in the film (Fig. 3b). When the concentration of PITBT-TPE was higher than 22.5 mM, the emission spectrum of the DPATPAN/PITBT-TPE blends were identical to PITBT-TPE alone and full quenching of DPATPAN emission was observed (Fig. S1b, ESI†). At 250 mM in PMMA, the energy migration process between DPATPAN molecules has been previously identified to occur via a FRET mechanism using time-resolved fluorescence anisotropy experiments.11 In this work, PITBT-TPE molecules act as energy traps. The FRET critical distance (R0) between DPATPAN and PITBT-TPE molecules, at 250 mM and 22.5 mM in PMMA respectively, was 4.49 nm as calculated from the UV-Vis absorption and PL emission data. The mean inter-molecular distance between DPATPAN and PITBT-TPE was calculated to be 1.63 nm at these concentrations. This distance was within the R0 value but larger than typical distances required for a Dexter transfer mechanism to be operative (see ESI† for calculation details).
 | ||
| Fig. 3 (a) ϕPL and (b) emission spectrum of DPATPAN/PITBT-TPE blends in PMMA. The concentration of DPATPAN in all mixtures was 250 mM while the concentration of PITBT-TPE was varied. The ϕPL of all samples were measured by an integrating sphere (see ESI† for details). Comparison of the normalized absorption and excitation spectra of (c) DPATPAN (250 mM), (d) DPATPAN (250 mM)/PITBT-TPE (90 mM) blend and (e) PITBT-TPE (90 mM) in PMMA. Spectra in (c and d) were normalized at maximum intensity, while spectra in (e) were normalized at 540 nm. | ||
We also observe consistently higher ϕPL of DPATPAN/PITBT-TPE blends when excited in the region where the donor strongly absorbs (390 nm) compared to direct excitation of the acceptor alone (Fig. 2b, 490 nm). We initially hypothesized that this observation was a result of a shift in dielectric environment from the addition of DPATPAN. The quantum yield was then measured by exciting the donor–acceptor blend at the acceptor's absorption maximum. In this case, the quantum yield (ϕPL = 57.8 ± 0.5%, PITBT-TPE 180 mM, 490 nm) returned to the same level of the acceptor alone at the same concentration (Fig. 2b), which suggested a quantum yield variation of the donor–acceptor blends against the excitation wavelength, rather than a variation in dielectric environment with increasing chromophore concentration. The ϕPL of the DPATPAN/PITBT-TPE blends in the PMMA matrix decreased gradually with increasing concentration of PITBT-TPE. We ascribed the decrease in the quantum yield of the DPATPAN/PITBT-TPE blends with increasing concentration of PITBT-TPE to concentration quenching of PITBT-TPE. Considering the emission of DPATPAN was fully quenched at 22.5 mM of PITBT-TPE and the ϕPL of the DPATPAN/PITBT-TPE blends were all higher than PITBT-TPE alone, the FRET efficiency from DPATPAN to PITBT-TPE should be close to unity. The difference between the absorption and excitation spectra measured from the DPATPAN (250 mM)/PITBT-TPE (90 mM) blend films can be ascribed to ϕPL variation with excitation wavelength in the DPATPAN/PITBT-TPE blends. The excitation and absorption spectra of the sample with DPATPAN only matched very well from 320 nm to 550 nm, which suggested that there was little variation in the ϕPL of DPATPAN in this wavelength range (Fig. 3c).
On the other hand, the excitation spectrum of the DPATPAN/PITBT-TPE blend was lower than its absorption spectrum from 400 nm to 540 nm, which suggested the ϕPL of the blend excited in this range was lower than at other wavelengths (Fig. 3d, spectra normalised at 540 nm). In addition, the excitation spectrum of PITBT-TPE was much lower than its absorption spectrum below 540 nm, which indicated the ϕPL of PITBT-TPE decreased when excited at wavelengths shorter than 540 nm (Fig. 3e, spectra normalised at 540 nm). These absorption and excitation spectra comparisons and the quantum yield data suggested a possible whereby resonance energy transfer from DPATPAN to PITBT-TPE avoids higher excitation energy loss in PITBT-TPE (see ESI† for an illustration of one possible mechanism, Fig. S3). Further studies are required to fully examine this effect and elucidate the mechanism.
The Monte Carlo ray tracing simulation (see ESI† for details) was carried out to further examine the optical properties of LSC devices based on a series of DPATPAN/PITBT-TPE fluorophore samples.18 In the simulation, the concentration of DPATPAN was maintained at 250 mM while the concentration of PITBT-TPE was at 22.5 mM, the minimum PITBT-TPE concentration at which the emission of DPATPAN was already fully quenched by FRET (Fig. 3b). As a reference, we also simulated the performance of an LSC that only contained 250 mM DPATPAN. The ϕPL of 250 mM DPATPAN in the presence or absence of 22.5 mM of PITBT-TPE were comparable (Fig. 3a). To compare the re-absorption effect of the DPATPAN-only and DPATPAN/PiTBT-TPE in a LSC device, we define the re-absorption value, FR, as:
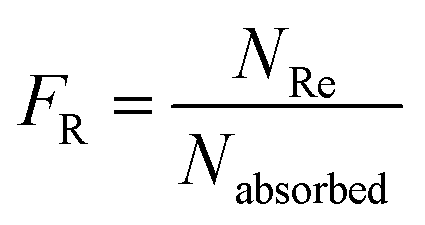
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000). As shown in Fig. 4, the simulated FR of the LSC that contained the DPATPAN/PITBT-TPE blend was lower than the LSC that only contained DPATPAN over the range of geometric gains that were simulated. This result shows that re-absorption was reduced in the DPATPAN/PITBT-TPE blend.
000). As shown in Fig. 4, the simulated FR of the LSC that contained the DPATPAN/PITBT-TPE blend was lower than the LSC that only contained DPATPAN over the range of geometric gains that were simulated. This result shows that re-absorption was reduced in the DPATPAN/PITBT-TPE blend.
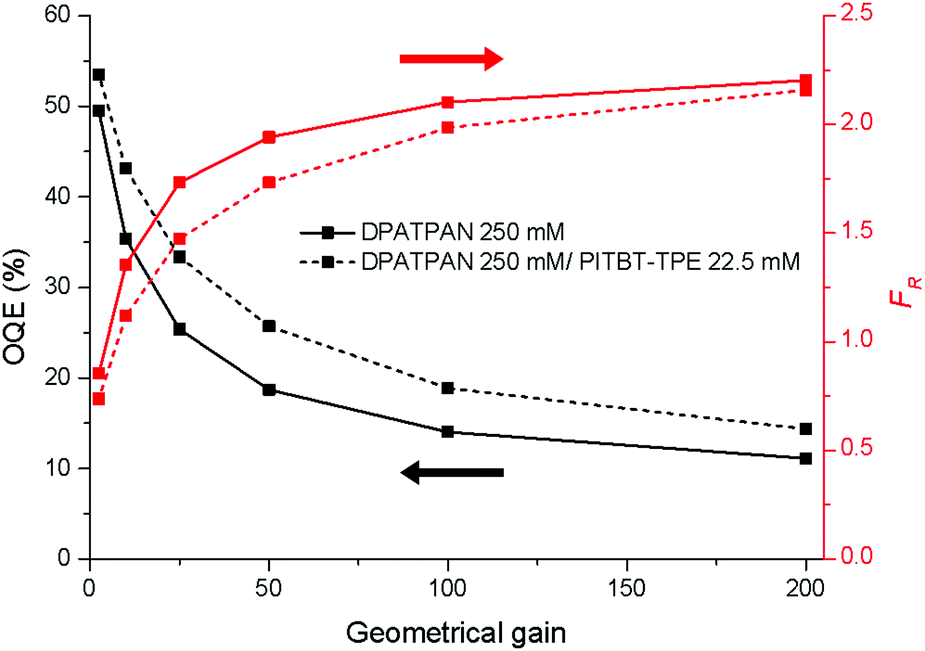 | ||
| Fig. 4 Simulated OQE (black curves) and re-absorption number (red curves) of the LSC devices as a function of the geometric gain. The devices were based on 250 mM DPATPAN with (dashed line) or without (continuous line) PITBT-TPE in a PMMA thin-film matrix casted on a 1 mm thick square glass waveguide. The absorbance of all samples was set to 1.0 at 390 nm. All samples were excited at 390 nm and a total of 100000 photons were traced in each simulation. | ||
The performance of the LSC devices was further characterised using optical quantum efficiency (OQE) as a comparison metric, define as:19
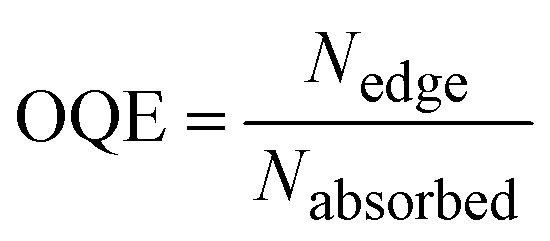
In conclusion, we have investigated the photophysics of PITBT-TPE in PMMA and its potential as an acceptor for LSCs that leverages energy migration and trapping to reduce reabsorption. It was observed that the quantum yield of PITBT-TPE can be sustained at high concentrations in PMMA. The blend composition of the DPATPAN/PITBT-TPE pair in thin-films has been optimised to maximize both the energy-transfer efficiency and emission quantum yield. Monte Carlo simulations of a LSC with optimal DPATPAN/PITBT-TPE composition predicted a reduction of re-absorption compared to LSCs with only DPATPAN. Our results provide insight and motivation into the development of AIE-based LSCs that can maximize light-harvesting as well as reduce reabsorption simultaneously.
Future work may additionally seek to broaden the absorption bandwidth of LSCs by using different AIE chromophores that will harvest most of the visible region of the solar spectrum and trap the absorbed energy to a low concentration, near-infrared emitter.20,21 Designing highly emissive dyes that can absorb most of the red region of the solar spectrum and also sustain high concentrations in thin-films22,23 are then desirable to achieve broadband light collection for LSCs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was made possible by support from the Australian Renewable Energy Agency which funds the project grants within the Australian Centre for Advanced Photovoltaics (ACAP). JLB was supported by an Australian Postgraduate Award and Eugen Singer Award during the period of this work. Responsibility for the views, information or advice expressed herein is not accepted by the Australian Government. WWHW is supported by an ARC Future Fellowship (FT130100500). KPG and WWHW are also supported by the ARC Centre of Excellence in Exciton Science (CE170100026). BZT acknowledges the Innovation and Technology Commission (ITC-CNERC14SC01) for support.References
- M. G. Debije, C. Tzikas, M. M. de Jong, M. Kanellis and L. H. Slooff, Renewable Energy, 2018, 116, 335–343 CrossRef
.
- M. G. Debije, C. Tzikas, V. A. Rajkumar and M. M. de Jong, Renewable Energy, 2017, 113, 1288–1292 CrossRef
- M. G. Debije and P. P. C. Verbunt, Adv. Energy Mater., 2012, 2, 12–35 CrossRef CAS
- M. Kanellis, M. M. de Jong, L. Slooff and M. G. Debije, Renewable Energy, 2017, 103, 647–652 CrossRef CAS
- B. McKenna and R. C. Evans, Adv. Mater., 2017, 29, 1606491 CrossRef PubMed
- C. J. Traverse, R. Pandey, M. C. Barr and R. R. Lunt, Nat. Energy, 2017, 2, 849–860 CrossRef
- A. Sanguineti, M. Sassi, R. Turrisi, R. Ruffo, G. Vaccaro, F. Meinardi and L. Beverina, Chem. Commun., 2013, 49, 1618–1620 RSC
- R. Turrisi, A. Sanguineti, M. Sassi, B. Savoie, A. Takai, G. E. Patriarca, M. M. Salamone, R. Ruffo, G. Vaccaro, F. Meinardi, T. J. Marks, A. Facchetti and L. Beverina, J. Mater. Chem. A, 2015, 3, 8045–8054 CAS
- J. L. Banal, K. P. Ghiggino and W. W. H. Wong, Phys. Chem. Chem. Phys., 2014, 16, 25358–25363 RSC
- J. L. Banal, H. Soleimaninejad, F. M. Jradi, M. Liu, J. M. White, A. W. Blakers, M. W. Cooper, D. J. Jones, K. P. Ghiggino, S. R. Marder, T. A. Smith and W. W. H. Wong, J. Phys. Chem. C, 2016, 120, 12952–12958 CAS
- J. L. Banal, B. Zhang, D. J. Jones, K. P. Ghiggino and W. W. H. Wong, Acc. Chem. Res., 2017, 50, 49–57 CrossRef CAS PubMed
- G. D. Gutierrez, I. Coropceanu, M. G. Bawendi and T. M. Swager, Adv. Mater., 2016, 28, 497–501 CrossRef CAS PubMed
- B. Zhang, H. Soleimaninejad, D. J. Jones, J. M. White, K. P. Ghiggino, T. A. Smith and W. W. H. Wong, Chem. Mater., 2017, 29, 8395–8403 CrossRef CAS
- Y. Gong, Y. Tan, J. Liu, P. Lu, C. Feng, W. Z. Yuan, Y. Lu, J. Z. Sun, G. He and Y. Zhang, Chem. Commun., 2013, 49, 4009–4011 RSC
- J. Zhou, B. He, J. Xiang, B. Chen, G. Lin, W. Luo, X. Lou, S. Chen, Z. Zhao and B. Z. Tang, ChemistrySelect, 2016, 1, 812–818 CrossRef CAS
- J. L. Banal, J. M. White, K. P. Ghiggino and W. W. H. Wong, Sci. Rep., 2014, 4, 4635 CrossRef PubMed
- L. R. Wilson, B. C. Rowan, N. Robertson, O. Moudam, A. C. Jones and B. S. Richards, Appl. Opt., 2010, 49, 1651–1661 CrossRef PubMed
- C. Haines, M. Chen and K. P. Ghiggino, Sol. Energy Mater. Sol. Cells, 2012, 105, 287–292 CrossRef CAS
- M. J. Currie, J. K. Mapel, T. D. Heidel, S. Goffri and M. A. Baldo, Science, 2008, 321, 226–228 CrossRef CAS PubMed
- R. Rondão, A. R. Frias, S. F. H. Correia, L. Fu, V. de Zea Bermudez, P. S. André, R. A. S. Ferreira and L. D. Carlos, ACS Appl. Mater. Interfaces, 2017, 9, 12540–12546 Search PubMed
- Y. Zhao, G. A. Meek, B. G. Levine and R. R. Lunt, Adv. Opt. Mater., 2014, 2, 606–611 CrossRef CAS
- F. De Nisi, R. Francischello, A. Battisti, A. Panniello, E. Fanizza, M. Striccoli, X. Gu, N. L. C. Leung, B. Z. Tang and A. Pucci, Mater. Chem. Front., 2017, 1, 1406–1412 RSC
- F. Gianfaldoni, F. De Nisi, G. Iasilli, A. Panniello, E. Fanizza, M. Striccoli, D. Ryuse, M. Shimizu, T. Biver and A. Pucci, RSC Adv., 2017, 7, 37302–37309 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qm00598a |
| This journal is © the Partner Organisations 2018 |