
Remarkable water-soluble ZnO nanocrystals: from ‘click’ functionalization to a supramolecular aggregation enhanced emission phenomenon†
Agnieszka
Grala
a,
Małgorzata
Wolska-Pietkiewicz
b,
Zbigniew
Wróbel
a,
Tomasz
Ratajczyk
a,
Joanna
Kuncewicz
c and
Janusz
Lewiński
 *ab
*ab
aInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: lewin@ch.pw.edu.pl
bFaculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland
cFaculty of Chemistry, Jagiellonian University, Gronostajowa 2, 30-387 Cracow, Poland
First published on 4th April 2018
Abstract
The preparation of ZnO nanocrystals with enhanced stability and photoluminescence intensity in an aqueous environment is a challenging task. We report on an one-pot two-step organometallic approach for the synthesis of processable alkyne-functionalized quantum-sized ZnO crystals and their efficient phase transfer protocol from an organic solvent to an aqueous environment involving the surface modification by the Cu-mediated azide–alkyne cycloaddition. Remarkably, the water-soluble NCs not only maintain the photoluminescence properties after the post-synthetic surface modification, but also exhibit the unprecedented (for II–VI semiconductor nanocrystals) aggregation enhanced emission phenomenon.
Introduction
The exceptional characteristics of semiconductor nanocrystals (NCs) make them pertinent as functional materials of diversified applicability in many areas of research, including optoelectronics, sensing, photovoltaics and bioimaging.1 While the spellbinding quantum-sized world has been dominated by cadmium-based NCs for the last two decades1,2 the potential practical and large-scale applications of these NCs are limited due to their negative biological and environmental impacts.3 Remarkably, for biomedical applications water solubilization of nanoparticles is also indispensable; however, the photoluminescence (PL) of semiconductor NCs is known to be often quenched in an aqueous environment.4ZnO-based nanostructures represent a highly desired alternative for heavy-metal based systems due to their biocompability5 and intrinsic physicochemical properties.6 For the past two decades, the inorganic sol–gel process has unceasingly played a key role in advancing the research on nanocrystalline ZnO forms. Nevertheless, this favorable inorganic method has several drawbacks, which extremely limit the NCs’ further use; for example, particularly substantial impediments are the low reproducibility and poor stability of ZnO quantum-sized crystals.7,8 Moreover, the resulting ZnO NCs are also not stable in a water environment and diverse surface modification strategies of the NCs’ water solubilization and stabilization have been developed.9 Due to the aforementioned impediments of the sol–gel method, the development of new efficient and reproducible synthetic methods affording well-defined and properly passivated quantum-sized ZnO crystals with enhanced stability and photoluminescence intensity in an aqueous environment has become highly desirable.
Greatly attractive and alternative methods to the inorganic sol–gel route are wet organometallic procedures. For example, Chaudret et al. elaborated the room-temperature synthesis of ZnO nanoparticles via controlled exposure to air of a homoleptic zinc alkyl precursor solution in the presence of long chain amines.10 More recently, Williams reported a new route for the synthesis of ZnO-based nanocomposites through the hydrolysis of a mixture of diethylzinc with substoichiometric quantities of inorganic zinc carboxylates or zinc phosphinates.11 Independently, we have developed two organometallic procedures affording high quality quantum-sized ZnO crystals which involve, respectively, (i) the self-supporting method based on the controlled exposure of the [EtZn(X)]n-type precursor to air leading to ZnO NCs with surface-anchored X-type ligands,12 and (ii) the hybrid method involving the direct hydrolysis of ZnEt2 along with the subsequent ZnO NCs’ surface decoration with selected X-type ligands.13 For example, we demonstrated that the latter two-step procedure provides NCs exhibiting ultra-long luminescence lifetimes with an average time constant of 2.2 μs for the slow component,12e,13 which is several orders of magnitude longer from that observed for ZnO nanostructures derived from the sol–gel process;14 these unique properties open prospects for both photovoltaic and photocatalytic applications. In turn, our ongoing studies show that the mentioned self-supporting organometallic approach produces bio-safe12e and highly processable ZnO NCs at an air/water interface,12a,b which can be subject to their first surface functionalization via Cu(I)-mediated click chemistry while retaining the PL properties of the NCs.8a
Advancing the self-supporting organometallic methodology, herein we report on an efficient phase transfer of alkyne-stabilized ZnO NCs from an organic solvent to water by their post-synthetic functionalization via Cu(I)-mediated click reaction. Strikingly, we revealed that the aliphatic chain length of the alkyne-capping ligand strongly affects the ZnO NCs’ solubilization. A combination of 5-hexynoic acid with hydrophilic 2-[2-(2-azidoethoxy)ethoxy]ethanol triggers the effective phase transfer process, during which the structure and optical properties of the ZnO NCs are well-protected, while analogues attempts involving 10-undecynoic carboxylate capping ligands failed. Moreover, water-soluble NCs not only maintain the PL and exhibit ultra-long-lived electron–hole separation after the post-synthetic surface modification, but also exhibit a spectacular PL enhancement upon the NCs’ supramolecular aggregation in an aqueous environment. We note that the aggregation-induced emission enhancement (AEE) and the related aggregation induced emission (AIE) phenomenon have hitherto been commonly associated with small non-luminescent in the solution state, but with high emission in aggregated or solid state organic molecules (AIEgens).15 Nowadays, attracted by these unique characteristics, many researchers have worked on the design and preparation of new types of AIEgens. However, to date there have been only a handful of examples of novel AIE systems including Ag/Au16 and Cu nanoclusters,17 Ga nanohybrids,18 and Cu–In–S quantum dots.19 And to the best of our knowledge, this is the first example of the AEE effect observed for II–VI quantum-sized semiconductor NCs.
Results and discussion
Clickable alkyne-functionalized ZnO-X NCs (where X = a deprotonated carboxylic acid with a terminal acetylene unit) were prepared using the recently developed one-pot two-step self-supporting organometallic approach.8a,12 For this purpose, two commercially available bifunctional carboxylic acids were selected as pro-ligands, namely 10-undecynoic acid (abbreviated as una-H) and 5-hexynoic acid (abbreviated as hex-H) (Scheme 1). Initially, [EtZn(X)]n-type precursors were synthesized in situ through the equimolar reaction of commercially available ZnEt2 with the selected X–H pro-ligands. In the second step, a solution of the corresponding ethylzinc precursor in THF was exposed to air to initiate transformations leading to ZnO NCs coated with the respective carboxylate X-type ligand shell (Scheme 1), further abbreviated as ZnO-una1 NCs and ZnO-hex1 NCs (for details, see the Experimental section). The average diameters of the resulting ZnO NCs were assessed from scanning transmission electron microscopy images, which were obtained from at least 100 NCs. The average diameters of the ZnO-una1 NCs and ZnO-hex1 NCs are 4.8 ± 0.5 nm (which is consistent with previously reported data8a) and 4.5 ± 0.5 nm, respectively (see Fig. 1a, b and Fig. S1–S3 in the ESI†). The inorganic core diameters nicely corroborated with the corresponding average hydrodynamic diameters of 8.7 nm8a and 5.6 nm (see Fig. S4a in the ESI†). The as-prepared ZnO NCs were additionally characterized using powder X-ray diffraction (PXRD), infrared spectroscopy (FTIR) and nuclear magnetic resonance (NMR) (for details, see the Experimental section and the ESI†).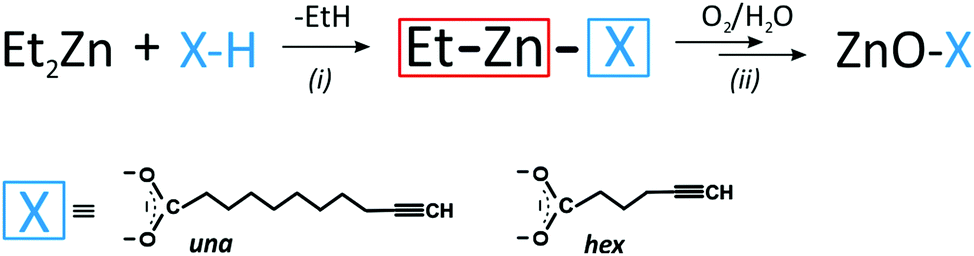 | ||
| Scheme 1 Schematic representation of (i) the synthesis of an [EtZn(X)]n-type precursor and (ii) its transformation into X-type carboxylate coated ZnO NCs. | ||
 | ||
| Fig. 1 STEM and HRTEM micrographs for (a and b) ZnO-hex1 NCs (DMSO), (c and d) ZnO-hex2 NCs (DMSO), and (e and f) ZnO-hex2 NCs (after 3 days in H2O). | ||
Subsequently, the surface post-synthetic modification of the resulting ZnO-X NCs via copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reaction was performed using a miniPEG derivative with OH termini (i.e. 2-[2-(2-azidoethoxy)ethoxy]ethanol solution, abbreviated as azOH) as a water-solubilizing modifier. Initially, we subjected ZnO-una1 NCs to the CuAAC reaction with azOH in THF. Remarkably, the 1H NMR spectrum of the post-synthetically modified ZnO-una1 NCs (ZnO-una2 NCs) contains a well-resolved signal characteristic for the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH moiety from unreacted una ligand (2.74 ppm) and sets of signals for the OH-terminated triazole molecule (Fig. S6 in the ESI†). This observation indicates that the click reaction led to the formation of ZnO-una2 NCs with a heterogeneous organic shell composed of both deprotonated carboxylic ligands and OH-terminated triazole derivatives (for details, see Fig. S6 and S8 in the ESI†). As estimated from the spectrum, approximately half of the una ligands from ZnO-una1 NCs reacted with azOH, and even the application of a two-fold excess of azide did not affect the conversion degree of the surface ligands. Disappointingly, the resulting miscellaneous surface capping layer precluded ZnO-una2 NCs’ phase transfer to the aqueous environment. It is worth nothing that the revealed degree of post-synthetic functionalization of ZnO-una1 NCs strongly contrasts with that observed in a previously reported click reaction involving ZnO-una1 NCs and S-(3-azidopropyl)thioacetate subunits which proceeded almost quantitatively.8a These data univocally demonstrate that the character of an azide subunit has a profound impact on the effectiveness of the post-synthetic functionalization of the NCs’ surfaces using the CuAAC reaction.
CH moiety from unreacted una ligand (2.74 ppm) and sets of signals for the OH-terminated triazole molecule (Fig. S6 in the ESI†). This observation indicates that the click reaction led to the formation of ZnO-una2 NCs with a heterogeneous organic shell composed of both deprotonated carboxylic ligands and OH-terminated triazole derivatives (for details, see Fig. S6 and S8 in the ESI†). As estimated from the spectrum, approximately half of the una ligands from ZnO-una1 NCs reacted with azOH, and even the application of a two-fold excess of azide did not affect the conversion degree of the surface ligands. Disappointingly, the resulting miscellaneous surface capping layer precluded ZnO-una2 NCs’ phase transfer to the aqueous environment. It is worth nothing that the revealed degree of post-synthetic functionalization of ZnO-una1 NCs strongly contrasts with that observed in a previously reported click reaction involving ZnO-una1 NCs and S-(3-azidopropyl)thioacetate subunits which proceeded almost quantitatively.8a These data univocally demonstrate that the character of an azide subunit has a profound impact on the effectiveness of the post-synthetic functionalization of the NCs’ surfaces using the CuAAC reaction.
Afterwards, to verify the effect of the aliphatic tail length of the supporting ligand on ZnO NCs’ phase transfer strategy, ZnO-hex1 NCs were subjected to the click reaction with azOH as a water-solubilizing modifier. In this case, the post-synthetic modification also led to the formation of a heterogeneous surface capping layer, but strikingly, the resulting ZnO-hex2 NCs were easily transferred to water. As shown in Fig. 2b, in the 1H NMR spectrum of ZnO-hex2 NCs, signals characteristic of the triazole species emerged, with the most distinctive resonance in the aromatic region at 7.81 ppm assignable to the ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–N moiety. Additionally, signals corresponding both to triazole and to deprotonated 5-hexynoic acid species were present (note that the CuAAC reaction proceeded in a similar manner for both una1- and hex1-coated ZnO NCs), which indicated that in this click reaction a similar heterogeneous organic layer was also formed (Scheme 2 and Fig. 2). The relating comparison of the 1H NMR spectra of the pure hex-H pro-ligand and ZnO-hex1 NCs are presented in Fig. S7 (ESI†). The FTIR data (Fig. S9, ESI†) substantiated the aforementioned 1H NMR analysis and confirmed the presence of both functionalities on the ZnO-hex2 NCs’ surfaces. Signals corresponding to CCH stretches at 3294 and 3277 cm−1 were significantly suppressed and intense bands at 1120 and 1064 cm−1 corresponding to the stretching vibrations of ether moieties appeared, which confirmed the presence of triazole rings. As anticipated, STEM and HRTEM micrographs demonstrated that the inorganic core mean diameters remained intact after the CuAAC reaction and for the ZnO-hex2 NCs dissolved in DMSO were of 4.7 ± 0.5 nm size (Fig. 1c, d and Fig. S3 in the ESI†), while the average hydrodynamic diameter was found to be much larger (9.18 nm) when compared to ZnO-hex1 NCs (see Fig. S4b in the ESI†). The PXRD patterns of both ZnO-hex1 and ZnO-hex2 NCs were consistent with a model pattern of the hexagonal wurtzite-type ZnO phase (see Fig. S10 in the ESI†) and the absence of impurities in the PXRD patterns confirms the high purity of the processed nanomaterial.
CH–N moiety. Additionally, signals corresponding both to triazole and to deprotonated 5-hexynoic acid species were present (note that the CuAAC reaction proceeded in a similar manner for both una1- and hex1-coated ZnO NCs), which indicated that in this click reaction a similar heterogeneous organic layer was also formed (Scheme 2 and Fig. 2). The relating comparison of the 1H NMR spectra of the pure hex-H pro-ligand and ZnO-hex1 NCs are presented in Fig. S7 (ESI†). The FTIR data (Fig. S9, ESI†) substantiated the aforementioned 1H NMR analysis and confirmed the presence of both functionalities on the ZnO-hex2 NCs’ surfaces. Signals corresponding to CCH stretches at 3294 and 3277 cm−1 were significantly suppressed and intense bands at 1120 and 1064 cm−1 corresponding to the stretching vibrations of ether moieties appeared, which confirmed the presence of triazole rings. As anticipated, STEM and HRTEM micrographs demonstrated that the inorganic core mean diameters remained intact after the CuAAC reaction and for the ZnO-hex2 NCs dissolved in DMSO were of 4.7 ± 0.5 nm size (Fig. 1c, d and Fig. S3 in the ESI†), while the average hydrodynamic diameter was found to be much larger (9.18 nm) when compared to ZnO-hex1 NCs (see Fig. S4b in the ESI†). The PXRD patterns of both ZnO-hex1 and ZnO-hex2 NCs were consistent with a model pattern of the hexagonal wurtzite-type ZnO phase (see Fig. S10 in the ESI†) and the absence of impurities in the PXRD patterns confirms the high purity of the processed nanomaterial.
 | ||
| Fig. 2 1H NMR spectra of (a) ZnO-hex1 NCs and (b) ZnO-hex2 NCs; * – DMSO-d6, a – water impurity, b – catalyst residue. | ||
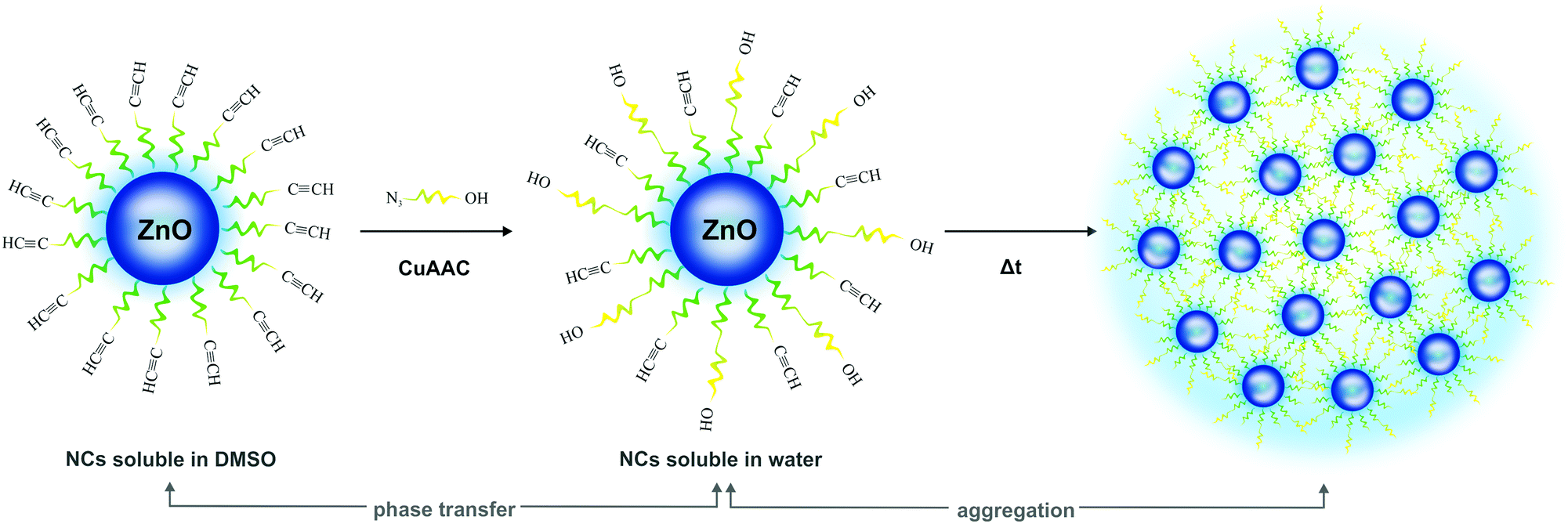 | ||
| Scheme 2 Graphical representation of the ZnO-hex1 NC functionalization via the CuAAC process leading to water-soluble ZnO-hex2 NCs and their tendency to aggregation accompanied by the PL enhancement. | ||
The cycloaddition reaction on the ZnO-hex1 NCs’ surfaces in the presence of the Cu(I) catalyst led only to a negligible photoluminescence decrease in DMSO (Fig. 3), which confirms that the organometallic approach leads to well-passivated ZnO NCs with an impermeable organic shell (the respective absorption spectra are shown in Fig. S11a in the ESI†). Then, spectroscopic studies were performed to determine the PL stability of ZnO-hex2 in a water environment under storage for four days (Fig. S12 in the ESI†). We have to point out that due to the high quality and impermeability of the organic ligand shell, a photoluminescence quenching effect4 in the aqueous environment (common for semiconductor nanostructures) was also not observed. Conversely, with time the emissive properties of ZnO-hex2 NCs were found to be significantly enhanced (Fig. S12, ESI†) and the solution became slightly turbid. Interestingly, the as-prepared ZnO-hex2 NCs after dissolution in water exhibit the time-dependent formation of aggregated structures (Fig. 1e, f and Fig. S2 in the ESI†), which are also visible to the naked eye. However, the inorganic core diameters of the ZnO-hex2 NCs dissolved in water did not change as presented in STEM and HRTEM micrographs (Fig. 1e, f and Fig. S2f–k, ESI†). As we recently showed, for carboxylate ligand-coated ZnO NCs the aggregation process at the air/water interface was driven by interdigitation of coating ligands instead of the inorganic core–core merging.12a,b Accordingly, it is reasonable to propose that in the case of ZnO-hex2 NCs the supramolecular aggregation is also driven by the NCs’ heterogeneous capping layers.20
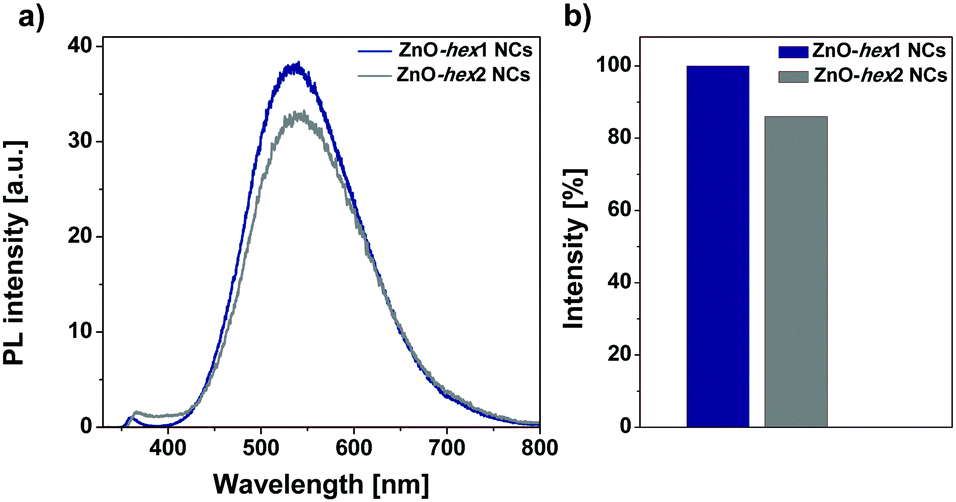 | ||
| Fig. 3 (a) PL emission spectra of ZnO-hex1 NCs (before the CuAAC reaction) and ZnO-hex2 NCs (after the CuAAC) in DMSO; and (b) the corresponding bar graphs representing the slight PL intensity suppression. PL intensities were measured with excitation at 330 nm. | ||
These observations prompted us to investigate the ZnO-hex2 NCs’ behavior in terms of the AEE phenomenon related to the poor photoluminescence of separate molecules in solution and their significantly stronger emission in the aggregated state.15,21 Strikingly, the emissivity of ZnO-hex2 was found to be strongly dependent on the water fraction in DMSO solution (Fig. 4). The most enhanced fluorescence intensity of two orders of magnitude was achieved in a solution containing 10% of DMSO and 90% of water (v/v). The PL dramatically increased and the maximum shifted from 555 nm to ∼539 nm (note that both PL peaks observed in Fig. 4 increased differently). The presence of water induced the aggregation of NCs, which was also confirmed by DLS analysis (see Fig. S5 in the ESI†). The addition of 50% of H2O (v/v) led to rapid diameter growth and after 24 h three ZnO NC populations were observed. In a solution containing 90% of H2O (v/v) NCs, the hydrodynamic diameters were even greater, and after 5 days three populations of ∼79, 122 and 220 nm diameters were found (see Fig. S5 in the ESI†).
 | ||
| Fig. 4 PL emission spectra of ZnO-hex2 NCs exhibiting the AEE phenomenon; i.e. 0% – pure DMSO, 90% – sample in 10% DMSO/90% H2O (v/v). PL intensities were measured with excitation at 330 nm. | ||
The AEE and AIE effects known for small organic molecules have been extensively explored for the last decade;15 however, to the best of our knowledge, these phenomena have not been recognized yet for II–VI semiconductor NCs. Our studies demonstrate that the post-synthetic ZnO NCs’ organic shell modification via conjugation with appropriate subunits induces supramolecular aggregation of the NCs, which in turn induces emission enhancement of the organic–inorganic core–shell type nanohybrid material. Nevertheless, at this stage of studies the mechanistic foundation behind the AEE phenomenon observed for ZnO-hex2 NCs in different DMSO–water mixture solvents is yet far from an explicit explanation and, undoubtedly, further research is demanded.
Additionally, for further characterization of ZnO-hex2 NCs the radiative recombination lifetimes were determined using time-resolved fluorescence spectroscopy. The decay profile for the NCs dispersed in DMSO revealed four components of the charge carrier dynamics, with two fast and two slow time constants of 18 ns, 54 ns, 0.45 μs and 1.30 μs, respectively. Analogously, for the ZnO-hex2 dispersed in a 50% DMSO/50% H2O (v/v) mixture also two fast and two slow decay components were observed with time constants of 5 ns, 20 ns, 0.13 μs and 0.45 μs, respectively (for details see Fig. S13 in the ESI†). As evidenced, for the NCs prepared using the self-supporting organometallic methodology the charge carrier lifetimes are significantly greater than typically observed for ZnO nanostructures derived from the sol–gel method for which luminescence decays of ∼10–20 ps were reported.14 Thus, these data are another instance of the superiority of the self-supporting organometallic procedure in the preparation of high quality ZnO quantum-sized crystals over the traditional sol–gel process.
Conclusions
In conclusion, we have successfully developed an efficient protocol for phase transfer of hydrophobic X-type ligand coated ZnO NCs from an organic solvent to water using the CuAAC reaction, i.e. one of the most frequently used click reactions for bioconjugation. Conversely to the commonly observed luminescence quenching of quantum-sized semiconductor crystals upon their aggregation in aqueous medium,4 the reported water-soluble ZnO-hex2 NCs exhibited both ultra-long-lived electron–hole separation and the unprecedented (for II–VI semiconductor nanomaterials) AEE effect dependent on the water fraction in DMSO solution. The reported results pave the way for countless types of colloidal semiconductor quantum-sized crystals as exceptional water-processable building blocks for the next generation of hybrid devices. Further studies on the effect of NCs’ organic shells on AEE and related AIE phenomena are in progress.Experimental
General considerations
The precursor synthesis and the click reaction on the surfaces of the NCs were conducted in an inert gas atmosphere using standard Schlenk techniques. THF, used as a solvent, was dried and distilled from a sodium–potassium alloy and benzophenone prior to use. All reagents were purchased from commercial vendors and used in a dry nitrogen atmosphere.Methods
The NMR spectra were acquired on a Bruker 300 MHz spectrometer at 298 K. The infrared spectra were recorded on a FTIR Tensor II Bruker spectrometer. Powder XRD data were collected on a Siemens D5005 diffractometer (Bruker AXS). Measurements employed Ni-filtered Cu Kα radiation of a copper sealed tube charged with 40 kV voltage and 40 mA current in a Bragg–Brentano geometry with a beam divergence of 1 deg. in the scattering plane. The sample was spread over the surface of a porous glass plate fixed to the sample holder. Diffraction patterns were measured in the scattering angle range of 10–80 degrees by step scanning in steps of 0.02 degree. The sizes and morphologies of the ZnO NCs were imaged using a Cs corrected scanning transmission electron microscope (STEM, Hitachi HD 2700, 200 kV). The scanning transmission electron microscopy (STEM) observations were carried out in three modes: SE, HAADF and HR TEM. Nanocrystal samples were drop-cast (DMSO solution) onto 300-mesh, holey carbon-coated copper grids (Quantifoil). Afterward, the excess solvent was evaporated at room temperature. Optical absorption (UV-vis) spectra for ZnO NC colloidal suspensions in DMSO, DMSO/H2O or H2O were collected on a Hitachi U-2910 spectrophotometer. A standard quartz cell (Hellma) with a 10 mm path length was used and rinsed with appropriate solvent before each run. Photoluminescence (PL) measurements were carried out using a Hitachi Fluorescence Spectrophotometer F-7000. The hydrodynamic diameter of NCs was determined by Dynamic Light Scattering (DLS) performed on a Malvern Zetasizer Nano Z (note that (a) freshly-prepared solutions of ZnO-hex1 NCs and ZnO-hex2 NCs in DMSO, DMSO/H2O or H2O were filtered before the DLS analysis through a 0.2 micron filter to remove any dust particles, (b) in the case of ZnO-hex2 solutions in DMSO/H2O or H2O we wait for the formation of aggregates and then perform DLS measurements). The PL lifetimes were measured at 20 °C using a single-photon counting system UV-VIS-NIR Fluorolog 3 Spectro-fluorimeter (Horiba Jobin Yvon). The solid-state pulsed NanoLED (λmax = 336 nm) was used as an excitation source. PL decay signals with a nanosecond resolution were obtained using a photomultiplier tube. The instrument response function was acquired with a LUDOX scatterer. The obtained luminescence decay curves were fitted using a four-exponential function.Synthesis of [EtZn(X)]n (where X = una or hex) and its transformation into ZnO-X NCs
(a) The procedure for [EtZn(una)]n synthesis and its transformation into ZnO-una1 NCs as well as ZnO-una1 NCs’ properties were previously reported.8a (b) [EtZn(hex)]n: 1.0 mmol of 5-hexynoic acid (112 mg) was kept in vacuo for 2 h before it was dissolved in THF (4 mL) and cooled to −78 °C in a nitrogen atmosphere. Then, ZnEt2 (0.5 mL of a 2 M solution in hexane, 1 mmol) was added dropwise under vigorous stirring. The reaction was gradually warmed up to room temperature. After 3 h the product [EtZn(hex)]n as a white solid was isolated, dried in vacuo and characterized spectroscopically. 1H NMR (C6D6, 300 MHz, 298 K): δ = 0.93 (q, ZnCH2CH3, 2H), 1.31 (m, –CH2C, 2H), 1.38 (t, ZnCH2CH3, 3H), 1.60 (t, –CH2–, 2H), 1.72 (s, CH, 1H), 1.89 + 2.29 (m, –CH2CO2–, 2H) ppm; 13C NMR (C6D6, 75 MHz, 298 K): δ =18.61, 25.06, 30.69, 36.50, 68.40, 70.02, 84.04, 188.51 ppm; IR (ATR): ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) = 3285 (vw), 2961 (vw), 2939 (vw), 2910 (vw), 2872 (vw), 2114 (vw), 2079 (vw), 1540 (vs), 1426 (s), 1408 (s), 1353 (w), 1320 (w), 1304 (w), 1274 (w), 1260 (w), 1233 (vw), 1161 (vw), 1097 (vw), 1056 (w), 1023 (w), 1016 (w), 896 (w), 872 (w), 856 (w), 795 (w), 632 (m), 538 (m), 439 (m) cm−1. (c) Preparation of ZnO-hex1 NCs: our synthetic protocol for preparing ZnO-hex1 NCs involves two steps conducted in a one-pot manner. In the first step, an organozinc precursor [EtZn(hex)]n was synthesized in situ through the equimolar reaction of ZnEt2 with the selected proligand (hex-H). ZnEt2 in hexane (0.5 mL of a 2 M solution in hexane, 1 mmol) was added dropwise to a solution of 1.0 mmol of 5-hexynoic acid (112 mg) in −78 °C in THF, and then the reaction mixture was allowed to warm to room temperature and stirred for 3 h. In the second step, a solution of the [EtZn(hex)]n precursor in THF was exposed to air to initiate transformations and stirred for 2 days at room temperature. Subsequently, the liquid residual was evaporated in vacuo. Then the ZnO-hex1 NCs were redispersed in THF and were precipitated using hexane, centrifuged, and washed again with hexane. The as-prepared ZnO-hex1 NCs were dried under vacuum and characterized. 1H NMR (DMSO-d6, 300 MHz, 298 K): δ = 1.63 (quint, –CH2–, 2H), 2.16 (t, –CH2C, 2H), 2.76 (s, CH, 1H), 3.38 (sbr, –CH2CO2–, 2H) ppm; 13C NMR (DMSO-d6, 75 MHz, 298 K): δ = 18.72, 26.23, 35.34, 72.48, 85.75, 179.60 ppm; IR (ATR): = 3293 (vw), 3273 (vw), 2957 (vw), 2940 (vw), 2911 (vw), 2874 (vw), 2114 (vw), 1591 (w), 1525 (vs), 1450 (m), 1411 (m), 1401 (m), 1358 (w), 1325 (vw), 1309 (vw), 1286 (vw), 1234 (vw), 1171 (vw), 1054 (vw), 1031 (vw), 962 (vw), 926 (vw), 847 (vw), 778 (vw), 742 (w), 677 (w), 650 (w), 641 (w), 476 (m) cm−1.
= 3285 (vw), 2961 (vw), 2939 (vw), 2910 (vw), 2872 (vw), 2114 (vw), 2079 (vw), 1540 (vs), 1426 (s), 1408 (s), 1353 (w), 1320 (w), 1304 (w), 1274 (w), 1260 (w), 1233 (vw), 1161 (vw), 1097 (vw), 1056 (w), 1023 (w), 1016 (w), 896 (w), 872 (w), 856 (w), 795 (w), 632 (m), 538 (m), 439 (m) cm−1. (c) Preparation of ZnO-hex1 NCs: our synthetic protocol for preparing ZnO-hex1 NCs involves two steps conducted in a one-pot manner. In the first step, an organozinc precursor [EtZn(hex)]n was synthesized in situ through the equimolar reaction of ZnEt2 with the selected proligand (hex-H). ZnEt2 in hexane (0.5 mL of a 2 M solution in hexane, 1 mmol) was added dropwise to a solution of 1.0 mmol of 5-hexynoic acid (112 mg) in −78 °C in THF, and then the reaction mixture was allowed to warm to room temperature and stirred for 3 h. In the second step, a solution of the [EtZn(hex)]n precursor in THF was exposed to air to initiate transformations and stirred for 2 days at room temperature. Subsequently, the liquid residual was evaporated in vacuo. Then the ZnO-hex1 NCs were redispersed in THF and were precipitated using hexane, centrifuged, and washed again with hexane. The as-prepared ZnO-hex1 NCs were dried under vacuum and characterized. 1H NMR (DMSO-d6, 300 MHz, 298 K): δ = 1.63 (quint, –CH2–, 2H), 2.16 (t, –CH2C, 2H), 2.76 (s, CH, 1H), 3.38 (sbr, –CH2CO2–, 2H) ppm; 13C NMR (DMSO-d6, 75 MHz, 298 K): δ = 18.72, 26.23, 35.34, 72.48, 85.75, 179.60 ppm; IR (ATR): = 3293 (vw), 3273 (vw), 2957 (vw), 2940 (vw), 2911 (vw), 2874 (vw), 2114 (vw), 1591 (w), 1525 (vs), 1450 (m), 1411 (m), 1401 (m), 1358 (w), 1325 (vw), 1309 (vw), 1286 (vw), 1234 (vw), 1171 (vw), 1054 (vw), 1031 (vw), 962 (vw), 926 (vw), 847 (vw), 778 (vw), 742 (w), 677 (w), 650 (w), 641 (w), 476 (m) cm−1.
CuAAC reaction on ZnO-X NCs’ surfaces
ZnO-X NCs prepared from 1 mmol of the [EtZn(X)]n precursor and 0.02 mmol of catalyst ([Cu(C12H8N2)[P(C6H5)3]2]NO3·1/2CH2Cl2) were placed in a Schlenk vessel and kept in vacuo for two hours before their solubilization in 2 mL of a THF solution. In another vessel with 2 mL of THF placed in a cooling bath (−20 °C) 1 mmol of 2-[2-(2-azidoethoxy)ethoxy]ethanol was added and after 30 min of cooling the mixture was exposed to vacuum in order to remove the oxygen (the flask was covered with aluminum foil to prevent the azide photodecomposition). Subsequently, the mixture was transferred with a syringe to the vessel with ZnO-X NCs (covered with aluminum foil), warmed up slowly to room temperature and stirred for 2 hours. After that time, the resulting product was isolated from the solution, washed two times with THF and dried in vacuo. We noted that even the application of a two-fold excess of azOH did not affect the conversion degree of the surface ligands. The as-prepared ZnO NCs were characterized.triazprod, 2H), 2.58 (t, –CH2Cuna, 2H), 2.74 (s, CHuna, 1H), 3.36 (m, –CH2Otriazprod–, 4H), 3.47 (m, –CH2Otriazprod–, 4H), 3.78 (t, –CH2OHtriazprod–, 2H), 4.45 (t, CH2Ntriazprod– 2H), 4.60 (t, –OHtriazprod–, 1H), 7.81 (s, CH–Ntriazprod–, 1H) ppm; una – signals assignable to una and triazprod – signals assignable to the triazole product bound to the surface; IR (ATR): = 3400 (vw), 3279 (vw), 3134 (vw), 2921 (w), 2849 (w), 2105 (vw), 1595 (vw), 1535 (vs), 1454 (m), 1396 (m), 1367 (vw), 1356 (vw), 1337 (vw), 1316 (vw), 1261 (vw), 1209 (vw), 1197 (vw), 1116 (w), 1101 (w), 1073 (w), 1034 (w), 950 (vw), 933 (vw), 917 (vw), 882 (vw), 849 (vw), 802 (vw), 797 (vw), 742 (w), 723 (w), 699 (w), 670 (w), 643 (w), 576 (vw), 550(vw), 461 (m) cm−1.
hex– + –CH2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/i_char_e001.gif) triazprod, 4H), 2.62 (sbr, –CH2CO2triazprod–, 2H), 2.72 (s, CHhex, 1H), 3.39 (m, –CH2CO2hex–, 2H), 3.48 (m, –CH2Otriazprod–, 4H), 3.51 (m, –CH2Otriazprod–, 4H), 3.79 (t, –CH2OHtriazprod–, 2H), 4.46 (t, CH2Ntriazprod– 2H), 4.55 (sbr, –OHtriazprod–, 1H), 7.81 (s, CH–Ntriazprod–, 1H) ppm; 13C NMR (DMSO-d6, 75 MHz, 298 K): δ = 18.82, 24.36, 26.13, 31.93, 50.45, 61.47, 70.01, 70.84, 72.46, 73.55, 85.73, 123.54, 129.93, 148.03, 179.80 ppm; hex – signals assignable to hex and triazprod – signals assignable to the triazole product bound to the surface; IR (ATR): = 3395 (vw), 3278 (vw), 3138 (vw), 3080 (vw), 2931 (vw), 2913 (vw), 2867 (w), 2109 (vw), 1590 (s), 1411 (s), 1348 (w), 1304 (w), 1258 (w), 1219 (w), 1199 (w), 1117 (m), 1059 (s), 922 (w), 909 (w), 882 (w), 849 (w), 819 (w), 813 (w), 726 (w), 695 (w), 645 (m), 451 (vs) cm−1.
triazprod, 4H), 2.62 (sbr, –CH2CO2triazprod–, 2H), 2.72 (s, CHhex, 1H), 3.39 (m, –CH2CO2hex–, 2H), 3.48 (m, –CH2Otriazprod–, 4H), 3.51 (m, –CH2Otriazprod–, 4H), 3.79 (t, –CH2OHtriazprod–, 2H), 4.46 (t, CH2Ntriazprod– 2H), 4.55 (sbr, –OHtriazprod–, 1H), 7.81 (s, CH–Ntriazprod–, 1H) ppm; 13C NMR (DMSO-d6, 75 MHz, 298 K): δ = 18.82, 24.36, 26.13, 31.93, 50.45, 61.47, 70.01, 70.84, 72.46, 73.55, 85.73, 123.54, 129.93, 148.03, 179.80 ppm; hex – signals assignable to hex and triazprod – signals assignable to the triazole product bound to the surface; IR (ATR): = 3395 (vw), 3278 (vw), 3138 (vw), 3080 (vw), 2931 (vw), 2913 (vw), 2867 (w), 2109 (vw), 1590 (s), 1411 (s), 1348 (w), 1304 (w), 1258 (w), 1219 (w), 1199 (w), 1117 (m), 1059 (s), 922 (w), 909 (w), 882 (w), 849 (w), 819 (w), 813 (w), 726 (w), 695 (w), 645 (m), 451 (vs) cm−1.
AEE study of ZnO-hex2 NCs
To study the solvent-induced aggregation effect of ZnO-hex2 NCs, 200 μL of NC solution (∼2.5 mg mL−1 stock solution in DMSO) and 1800 μL of an appropriate DMSO/water mixture were added to a glass tube, vigorously stirred and then transferred to a quartz cuvette for PL measurements.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Foundation for Polish Science Team Program co-financed by the European Union under the European Regional Development Fund TEAM/2016-2/14 and the National Science Centre (Grant OPUS 2014/13/B/ST5/04420) for financial support. We thank Dr J. Grzonka (Faculty of Materials Science and Engineering, Warsaw University of Technology and Institute of Physical Chemistry Polish Academy of Sciences) for assistance with STEM imaging.Notes and references
- (a) F. Seker, K. Meeker, T. F. Kuech and A. B. Ellis, Chem. Rev., 2000, 100, 2505 CrossRef CAS PubMed; (b) A. M. Smith and S. Nie, Acc. Chem. Res., 2010, 43, 190 CrossRef CAS PubMed; (c) D. V. Talapin, J. S. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389 CrossRef CAS PubMed; (d) P. V. Kamat, K. Tvrdy, D. R. Baker and J. G. Radich, Chem. Rev., 2010, 110, 6664 CrossRef CAS PubMed; (e) J. Zhou, Y. Yang and C. Zhang, Chem. Rev., 2015, 115, 11669 CrossRef CAS PubMed.
- (a) H. Weller, Angew. Chem., Int. Ed. Engl., 1993, 32, 41 CrossRef; (b) C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706 CrossRef CAS; (c) A. P. Alivasatos, Science, 1996, 271, 933 Search PubMed.
- E. Oh, R. Liu, A. Nel, K. B. Gemill, M. Bilal, Y. Cohen and I. M. Medintz, Nat. Nanotechnol., 2016, 11, 479 CrossRef CAS PubMed.
- M. Noh, T. Kim, H. Lee, C.-K. Kim, S.-W. Joo and K. Lee, Colloids Surf., A, 2010, 359, 39 CrossRef CAS.
- (a) J. Zhou, N. S. Xu and Z. L. Wang, Adv. Mater., 2006, 18, 2432 CrossRef CAS; (b) Z. Li, R. Yang, M. Yu, F. Bai, C. Li and Z. L. Wang, J. Phys. Chem. C, 2008, 112, 20114 CrossRef CAS; (c) H.-M. Xiong, Adv. Mater., 2013, 25, 5329 CrossRef CAS PubMed.
- (a) C. D. Look, Mater. Sci. Eng., B, 2001, 80, 383 CrossRef; (b) U. Ozgur, Y. I. Alivov, C. Liu, A. Teke, M. A. Reshchikov, S. Dogan, U. Avrutin, S.-J. Cho and H. Morkoc, J. Appl. Phys., 2005, 98, 041301 CrossRef; (c) A. B. Djurišić and Y. H. Leung, Small, 2006, 2, 944 CrossRef PubMed; (d) C. Klingshirn, ChemPhysChem, 2007, 8, 782 CrossRef CAS PubMed.
- For selected examples, see: (a) M. S. Tokumoto, S. H. Pulcinelli, C. V. Santilli and V. Briois, J. Phys. Chem. B, 2003, 107, 568 CrossRef CAS; (b) M. Voigt, M. Klaumunzer, H. Thiem and W. Peukert, J. Phys. Chem. C, 2010, 114, 6243 CrossRef CAS; (c) B. L. Caetano, C. V. Santilli, F. Meneau, V. Briois and S. H. Pulcinelli, J. Phys. Chem. C, 2011, 115, 4404 CrossRef CAS; (d) A. Layek, G. Mishra, A. Sharma, M. Spasova, S. Dhar, A. Chowdhury and R. A. Bandyopadhyaya, J. Phys. Chem. C, 2012, 116, 24757 CrossRef CAS; (e) T. Schindler, J. Walter, W. Peukert, D. Segets and T. Unruh, J. Phys. Chem. B, 2015, 119, 15370 CrossRef CAS PubMed.
- (a) A. Grala, M. Wolska-Pietkiewicz, W. Danowski, Z. Wróbel, J. Grzonka and J. Lewiński, Chem. Commun., 2016, 52, 7340 RSC; (b) For a discussion of different factors that influence the shielding of encapsulated fluorescent quantum dots against the penetration of copper(II) ions, see: J.-P. Merkl, J. Ostermann, C. Schmidtke, H. Kloust, R. Eggers, A. Feld, C. Wolter, A.-M. Kreuziger, S. Flessau, H. Mattoussi and H. Weller, in Colloidal Nanoparticles for Biomedical Applications IX, ed. W. J. Parak, J. M. Osinski, K. I. Yamamoto, Proc. of SPIE 8955, 2014, vol. 8955, p. 89551X Search PubMed.
- (a) R. A. Sperling and W. J. Parak, Philos. Trans. R. Soc., A, 2010, 368, 1333 CrossRef CAS PubMed; (b) R. O. Moussodia, L. Balan, C. Merlin, C. Mustin and R. Schneider, J. Mater. Chem., 2010, 20, 1147 RSC; (c) R. Mout, D. F. Moyano, S. Rana and V. M. Rotello, Chem. Soc. Rev., 2012, 41, 2539 RSC; (d) H.-J. Zhang, H.-M. Xiong, Q.-G. Ren, Y.-Y. Xia and J.-L. Kong, J. Mater. Chem., 2012, 22, 13159 RSC; (e) A. Dazzazi, Y. Coppel, M. In, C. Chassenieux, P. Mascalchi, L. Salome, A. Bouhaouss, M. L. Kahn and F. Gauffre, J. Mater. Chem. C, 2013, 1, 2158 RSC; (f) G. Palui, F. Aldeek, W. T. Wang and H. Mattoussi, Chem. Soc. Rev., 2015, 44, 193 RSC.
- (a) M. Monge, M. L. Kahn, A. Maisonnat and B. Chaudret, Angew. Chem., 2003, 42, 5321 CrossRef CAS PubMed; (b) M. L. Kahn, M. Monge, V. Collière, F. Senocq, A. Maisonnat and B. Chaudret, Adv. Funct. Mater., 2005, 15, 458 CrossRef CAS.
- (a) K. L. Orchard, M. S. P. Shaffer and C. K. Williams, Chem. Mater., 2012, 24, 2443 CrossRef CAS; (b) N. J. Brown, J. Weiner, K. Hellgardt, M. S. P. Shaffer and C. K. Williams, Chem. Commun., 2013, 49, 11074 RSC; (c) N. J. Brown, A. García-Trenco, J. Weiner, E. R. White, M. Allinson, Y. Chen, P. P. Wells, E. K. Gibson, K. Hellgardt, M. S. P. Shaffer and C. K. Williams, ACS Catal., 2015, 5, 2895 CrossRef CAS; (d) S. Noimark, J. Weiner, N. Noor, E. Allan, C. K. Williams, M. S. P. Shaffer and I. P. Parkin, Adv. Funct. Mater., 2015, 25, 1367 CrossRef CAS; (e) S. K. Sehmi, S. Noimark, S. D. Pike, J. C. Bear, W. J. Peveler, C. K. Williams, M. S. P. Shaffer, E. Allan, I. P. Parkin and A. J. MacRobert, ACS Omega, 2016, 1, 334 CrossRef CAS PubMed; (f) S. D. Pike, E. R. White, M. S. P. Shaffer and C. K. Williams, Nat. Commun., 2016, 7, 13008 CrossRef CAS PubMed.
- (a) J. Paczesny, M. Wolska-Pietkiewicz, I. Binkiewicz, Z. Wróbel, M. Wadowska, K. Matuła, I. Dzięcielewski, D. Pociecha, J. Smalc-Koziorowska, J. Lewiński and R. Hołyst, Chem. – Eur. J., 2015, 21, 16941 CrossRef CAS PubMed; (b) J. Paczesny, M. Wolska-Pietkiewicz, I. Binkiewicz, M. Wadowska, Z. Wróbel, K. Matuła, W. Nogala, J. Lewiński and R. Hołyst, ACS Appl. Mater. Interfaces, 2016, 8, 13532 CrossRef CAS PubMed; (c) M. Wolska-Pietkiewicz, A. Grala, I. Justyniak, D. Hryciuk, M. Jędrzejewska, J. Grzonka, K. J. Kurzydłowski and J. Lewiński, Chem. – Eur. J., 2017, 49, 11856 CrossRef PubMed; (d) E. Chwojnowska, M. Wolska-Pietkiewicz, J. Grzonka and J. Lewinski, Nanoscale, 2017, 9, 14786 RSC; (e) M. Wolska-Pietkiewicz, K. Tokarska, A. Grala, A. Wojewódzka, E. Chwojnowska, J. Grzonka, P. Cywiński, K. Kruczała, Z. Sojka, M. Chudy and J. Lewiński, Chem. – Eur. J., 2018, 24, 4033 CrossRef CAS PubMed.
- (a) A. M. Cieślak, M. V. Pavliuk, L. D’Amario, M. Abdellah, K. Sokołowski, U. Rybinska, D. L. A. Fernandes, M. Leszczyński, F. Mamedov, A. M. El-Zhory, J. Fohlinger, A. Budinskà, M. Wolska-Pietkiewicz, L. Hammarstrom, J. Lewiński and J. Sà, Nano Energy, 2016, 30, 187 CrossRef; (b) M. V. Pavliuk, A. M. Cieślak, M. Abdellah, A. Budinskà, S. Pullen, K. Sokołowski, D. L. A. Fernandes, J. Szlachetko, E. L. Bastos, S. Ott, L. Hammarstöm, T. Edvinsson, J. Lewiński and J. Sà, Sustainable Energy Fuels, 2017, 1, 69 RSC; (c) A. M. Cieślak, E. R. Janeček, K. Sokołowski, T. Ratajczyk, M. K. Leszczyński, O. A. Scherman and J. Lewiński, Nanoscale, 2017, 9, 16128 RSC.
- (a) T. J. Jacobsson, S. Viarbitskaya, E. Mukhtar and T. Edvinsson, Phys. Chem. Chem. Phys., 2014, 16, 13849 RSC; (b) Y. Zhong, A. B. Djurisic, Y. F. Hsu, K. S. Wong, G. Brauer, C. C. Ling and W. K. Chan, J. Phys. Chem. C, 2008, 112, 16286 CrossRef CAS; (c) L. Spanhel and M. A. Anderson, J. Am. Chem. Soc., 1991, 113, 2826 CrossRef CAS; (d) E. A. Meulenkamp, J. Phys. Chem. B, 1998, 102, 5566 CrossRef CAS.
- For selected reviews, see: (a) A. Qin, J. W. Y. Lam and B. Z. Tang, Prog. Polym. Sci., 2012, 37, 182 CrossRef CAS; (b) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS PubMed; (c) B. Yang, X. Zhang, Z. Zhang, Z. Huang, Y. Wei and L. Tao, Mater. Today, 2016, 19, 284 CrossRef CAS; (d) S. Chen, H. Wang, Y. Hong and B. Z. Tang, Mater. Horiz., 2016, 3, 283 RSC; (e) Y. Wang, G. Zhang, M. Gao, Y. Cai, C. Zhan, Z. Zhao, D. Zhang and B. Z. Tang, Faraday Discuss., 2017, 196, 9 RSC.
- (a) Z. Luo, X. Yuoan, Y. Yu, Q. Zhang, D. T. Leong, J. Y. Lee and J. Xie, J. Am. Chem. Soc., 2012, 134, 16662 CrossRef CAS PubMed; (b) X. Dou, X. Yuan, Y. Yu, Z. Luo, Q. Yao, D. T. Leong and J. Xie, Nanoscale, 2014, 6, 157 RSC; (c) Y. Yu, Z. Luo, D. M. Chevrier, D. T. Leong, P. Zhang, D.-E. Jiang and J. Xie, J. Am. Chem. Soc., 2014, 136, 1246 CrossRef CAS PubMed; (d) K. Zheng, X. Yuan, K. Kuah, Z. Luo, Q. Yao, Q. Zhang and J. Xie, Chem. Commun., 2015, 51, 15165 RSC; (e) R. Tian, S. Zhang, M. Li, Y. Zhou, B. Lu, D. Yan, M. Wei, D. G. Evans and X. Duan, Adv. Funct. Mater., 2015, 25, 5006 CrossRef CAS; (f) S. Jin, W. Liu, D. Hu, X. Zou, X. Kang, W. Du, S. Chen, S. Wei, S. Wang and M. Zhu, Chem. – Eur. J., 2018, 24, 3712 CrossRef CAS PubMed.
- X. Jia, X. Yang, J. Li, D. Li and E. Wang, J. Phys. Chem. Lett., 2016, 7, 962 CrossRef PubMed.
- T. Zhang, J.-L. Xu, X.-J. Wang, J. Zhang, X. Jiao, T. Wang and D. Chen, Chem. Commun., 2016, 52, 6922 RSC.
- M. Mou, Y. Wu, Q. Niu, Y. Wang, Z. Yan and S. Liao, Chem. Commun., 2017, 53, 3357 RSC.
- For the factors controlling the self-organization of nanoparticles, see: (a) E. V. Shevchenko, D. V. Talapin, N. A. Kotov, S. O’Brien and C. B. Murray, Nature, 2006, 439, 55 CrossRef CAS PubMed; (b) K. J. M. Bishop, C. E. Wilmer, S. Soh and B. A. Grzybowski, Small, 2009, 5, 1600 CrossRef CAS PubMed; (c) M. Grzelczak, J. Vermant, E. M. Furst and M. Liz-Marzàn, ACS Nano, 2010, 4, 3591 CrossRef CAS PubMed.
- (a) B.-K. An, S.-K. Kwon, S.-D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410 CrossRef CAS PubMed; (b) S. Shin, S. H. Gihm, C. R. Park, S. Kim and S. Y. Park, Chem. Mater., 2013, 15, 3288 CrossRef; (c) A. K. Sristava, A. Singh and L. Mishra, J. Phys. Chem. B, 2016, 120, 4490 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional STEM and HRTEM analysis, size distribution of ZnO NCs, dynamic light scattering, NMR spectroscopy, FT-IR spectroscopy, powder X-ray diffraction studies, UV-vis spectroscopy and PL lifetime measurements. See DOI: 10.1039/c7qm00586e |
| This journal is © the Partner Organisations 2018 |