
Synthesis of bithiazole-based semiconducting polymers via Cu-catalysed aerobic oxidative coupling†
Alanna
Faradhiyani
a,
Qiao
Zhang
 a,
Keisuke
Maruyama
a,
Junpei
Kuwabara
*a,
Takeshi
Yasuda
b and
Takaki
Kanbara
*a
a,
Keisuke
Maruyama
a,
Junpei
Kuwabara
*a,
Takeshi
Yasuda
b and
Takaki
Kanbara
*a
aTsukuba Research Center for Energy Materials Science (TREMS), Graduate School of Pure and Applied Sciences, University of Tsukuba, 1-1-1 Tennodai, Tsukuba, Ibaraki 305-8573, Japan. E-mail: kuwabara@ims.tsukuba.ac.jp; kanbara@ims.tsukuba.ac.jp
bResearch Center for Functional Materials, National Institute for Materials Science (NIMS), 1-2-1 Sengen, Tsukuba, Ibaraki 305-0047, Japan
First published on 3rd April 2018
Abstract
Polycondensation using Cu-catalysed aerobic oxidative C–H/C–H coupling reaction was developed for the synthesis of semiconducting polymers. The synthesised polymers served as optoelectronic materials in organic field effect transistors and organic light emitting diodes.
Conjugated polymers are promising semiconducting materials in organic electronic devices such as organic field effect transistors (OFETs), organic light emitting diodes (OLEDs), and organic photovoltaics.1–4 The recent developments in conjugated polymer materials have led to high-performance organic devices with practical applications.5 The improvement of synthetic methods for conjugated polymer materials is an important challenge to satisfy the demands of production costs, environmental issues, and quality of materials (molecular weight, purity, and a defect-free structure) for wide-scale practical applications.6 In recent years, polycondensation via direct C–H arylation has been investigated as an alternative synthetic method to conventional cross-coupling-based polycondensation.7–10 Direct arylation polycondensation is a facile strategy to reduce the number of synthetic steps for the monomers, and obtain highly pure polymers. This is because direct arylation polycondensation does not require organometallic monomers, and consequently no metal-containing by-products are formed.11,12 Although this method is a promising and practical approach to the production for conjugated polymer materials, a new strategy that allows for a further drastic improvement is still desired. One such ideal synthetic methods involves polycondensation by using an aerobic oxidative C–H/C–H coupling reaction.13 In this approach, the preparation of metalated and halogenated monomers can be avoided, and conjugated polymers without metal and halogen terminals, which are known to decrease the semiconducting performances, can be easily obtained.14–16 Furthermore, the utilization of dioxygen as a solo oxidant leads to a cost- and environmentally-friendly process because of the naturally abundant feedstock of dioxygen and harmless by-products such as H2O. Although polycondensation reactions using aerobic oxidative coupling of aromatic monomers have been previously reported in the preparation of conjugated polymers,17 there is scope for improvement in terms of reducing the amount of co-oxidant,18,19 circumventing the use of precious metal catalysts,20,21 and avoiding strong acidic conditions.22,23 This study aimed to develop an aerobic oxidative polycondensation using a Cu catalyst and dioxygen as a solo oxidant under neutral conditions. In addition, a site-selective direct arylation reaction was employed in monomer synthesis to increase the generality of this synthetic strategy, and to reduce the number of synthetic steps (Scheme 1). This method is able to afford bithiazole-based polymers, which serve as semiconducting materials in organic electronic devices.24–27 In this study, OFET and OLED properties of the synthesized polymers were evaluated to prove that the method is applicable to the preparation of semiconducting materials.
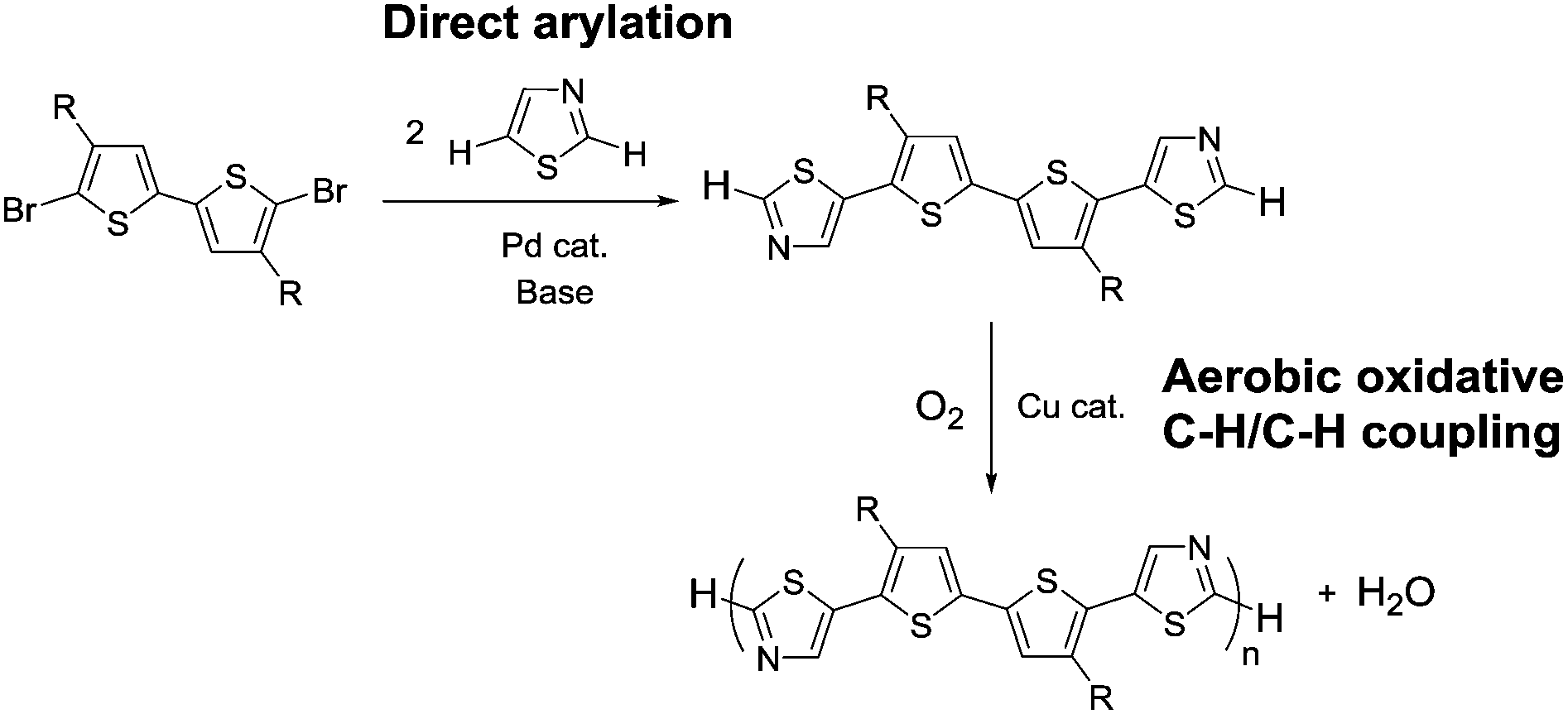 | ||
| Scheme 1 Synthetic strategy developed in this work, involving monomer synthesis via direct arylation at the C5 position of thiazole and aerobic oxidative polycondensation at the C2 position. | ||
A thiazole-based compound was selected as a target monomer because the acidic C–H bond at the 2-position in thiazole is reactive toward aerobic oxidative homo-coupling reactions.13,28–30 For synthesis of the target monomer, direct C–H arylation of thiazole at the 5-position was utilized in a selective manner because the C–H bond at the 5-position possesses a high reactivity in Pd-catalysed direct arylation (Scheme 1).31,32 Recently, Guo et al. reported a similar strategy of direct arylation for the synthesis of thiazole-based monomers and demonstrated the oxidative polymerization of the monomer using a stoichiometric amount of metal oxidants (Ag2CO3 or Cu(OAc)2).33 In this work, model monomers bearing a fluorene unit (M1 and M2) were selected for optimizing the reaction conditions and for a detailed structural analysis because of the expected high solubility of the corresponding polymers (Scheme 2). The direct arylation of thiazoles with a dibromofluorene derivative afforded the products in good yields in the presence of only 2 mol% of Pd(OAc)2 as a catalyst.32 These compounds were fully characterized by NMR, mass spectrometry, and elemental analysis.
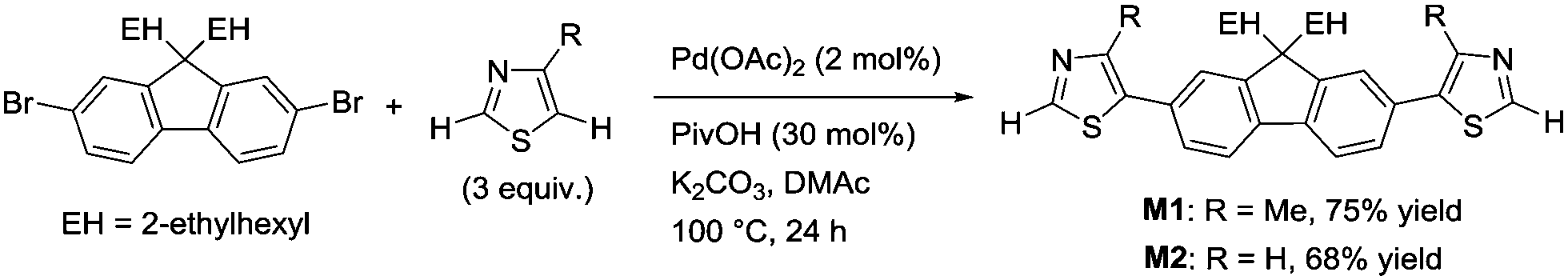 | ||
| Scheme 2 Synthesis of the monomers via C5-selective direct arylation of thiazoles. | ||
The monomer with methyl groups in the thiazole units (M1) was first examined for aerobic oxidative polymerization. The reaction with Cu(OAc)2 as a catalyst under air afforded a polymer with a molecular weight of 38![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 in 87% yield (Table 1, entry 1).29 The large polydispersity value (Mw/Mn = 4.48) might be caused by precipitation of polymeric products during polymerization. The polymerization of M2 also proceeded under the same reaction conditions (entry 2). The molecular weight of the P2 polymer (18500) was lower than that of the methyl substituted polymer because it precipitated during the reaction owing to its lower solubility.34 The reaction with a reduced amount of Cu(OAc)2 (10 mol%) afforded only oligomeric products (entry 3). A higher monomer concentration (0.5 M) increased the efficiency of the reaction, affording a high-molecular-weight polymer (entry 4). Reactions performed using an O2-filled balloon were less effective than those using an open system under air in terms of yields, molecular weight of the polymer, and reproducibility (Tables S1 and S2, ESI†). The presence of acid or base inhibited the reactions, although these additives reportedly promote the re-oxidation of the metal catalyst or contribute to the deprotonation step (Table S1, ESI†).35,36 Since the reaction with 5 mol% of the Cu catalyst afforded only oligomeric products, the reaction conditions in entry 4 have been determined as the optimized conditions.
500 in 87% yield (Table 1, entry 1).29 The large polydispersity value (Mw/Mn = 4.48) might be caused by precipitation of polymeric products during polymerization. The polymerization of M2 also proceeded under the same reaction conditions (entry 2). The molecular weight of the P2 polymer (18500) was lower than that of the methyl substituted polymer because it precipitated during the reaction owing to its lower solubility.34 The reaction with a reduced amount of Cu(OAc)2 (10 mol%) afforded only oligomeric products (entry 3). A higher monomer concentration (0.5 M) increased the efficiency of the reaction, affording a high-molecular-weight polymer (entry 4). Reactions performed using an O2-filled balloon were less effective than those using an open system under air in terms of yields, molecular weight of the polymer, and reproducibility (Tables S1 and S2, ESI†). The presence of acid or base inhibited the reactions, although these additives reportedly promote the re-oxidation of the metal catalyst or contribute to the deprotonation step (Table S1, ESI†).35,36 Since the reaction with 5 mol% of the Cu catalyst afforded only oligomeric products, the reaction conditions in entry 4 have been determined as the optimized conditions.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R | Cat./mol% | Conc./M | Yield/% | M n | M w/Mn |
| a The products were obtained by reprecipitation from CHCl3/CH3OH. Values of molecular weight were estimated by GPC calibrated on polystyrene standards. | ||||||
| 1 | CH3 | 20 | 0.25 | 87 | 38500 |
4.48 |
| 2 | H | 20 | 0.25 | 91 | 18500 |
4.66 |
| 3 | H | 10 | 0.25 | — | — | — |
| 4 | H | 10 | 0.50 | 98 | 19800 |
3.73 |
Fig. 1 shows the 1H NMR spectra of M2 and P2 synthesized under conditions outlined in entry 4. The signal for Ha in the monomer disappeared in the spectrum of the polymer because of the small number of the terminal unit in the high-molecular-weight polymer. All of the other signals could be assigned to protons in the repeating unit of the polymer with a reasonable integral ratio (Fig. S11, ESI†). These results show no defect of either branching or cross-linking structure in P2, although there is a possibility of a reaction at the 4-position of the thiazole moiety. The defect-free structure was also confirmed by 13C{1H} NMR (Fig. S12, ESI†). Inductively coupled plasma-mass spectrometry (ICP-MS) revealed that the residual amounts of Cu in P1 and P2 were 138 and 32 ppm, respectively. Therefore, the developed method was able to afford defect-free polymers with bench-stable Cu(OAc)2 as a catalyst and air as the sole oxidant source in a non-purified solvent. This simple reaction system is beneficial in terms of green chemistry.6,37
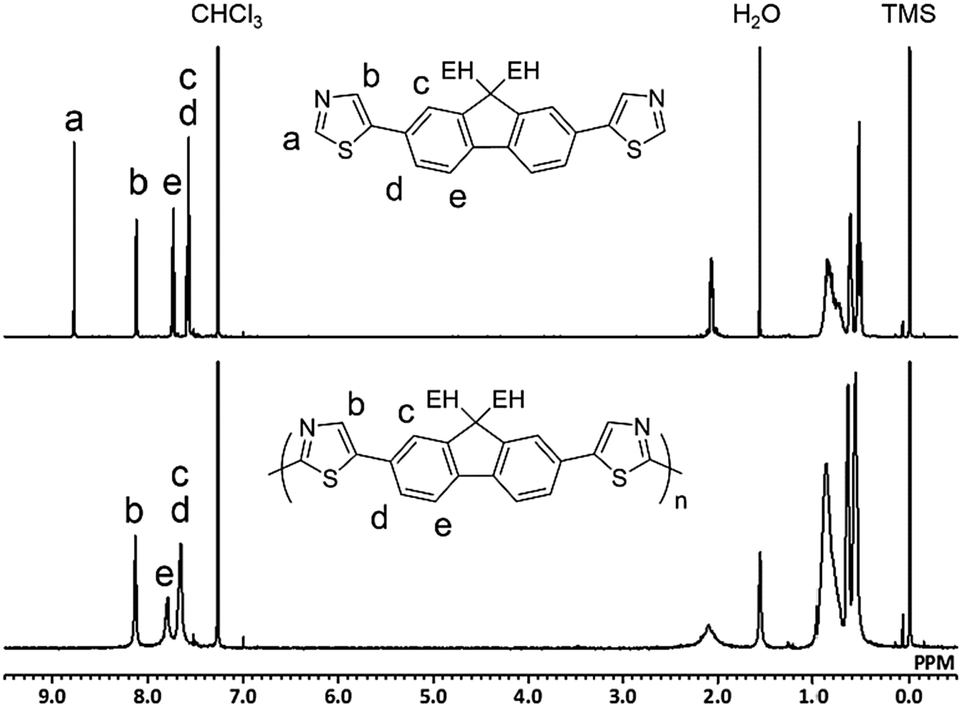 | ||
| Fig. 1 1H NMR spectra of M2 (top) and P2 (bottom) (CDCl3, 400 MHz). | ||
To prove the applicability of the developed method in the synthesis of semiconducting materials, the current method was applied to the synthesis of carbazole- and bithiophene-containing polymers. The monomers were synthesized by a similar direct arylation strategy under modified reaction conditions (Scheme S1, ESI†). The aerobic oxidative polycondensation of the monomers afforded the corresponding polymers (P3 and P4) under the optimized reaction conditions (Fig. 2). Owing to their low solubility, the polymers were obtained by Soxhlet extraction with o-dichlorobenzene. P3 and P4 were obtained in 80% and 73% yield, respectively. 1H NMR spectra of the polymers exhibit signals of the terminal unit (Fig. S13 and 14, ESI†). Based on the integral ratio between the terminal and repeating units, the degrees of polymerization of P3 and P4 were calculated to be 6 (MNMRn = 3400) and 8 (MNMRn = 5300), respectively. A high temperature GPC measurement for P4 exhibited a similar value (MGPCn = 5100), whereas that for P3 exhibited an underestimated value (MGPCn = 1100) presumably due to adsorption. The low molecular weights of P3 and P4 were likely a result of their precipitation during polymerization owing to low solubility. The detailed structural analyses proved the defect-free structure (see ESI†). Owing to relatively low-molecular weight, the terminal C–H groups were observed in 1H NMR spectra. ICP-AES showed a small amount of residual Cu (32 ppm for P3 and below measurable limits for P4).
 | ||
| Fig. 2 Structures of P3 and P4. | ||
The physical properties of P1–P4 were evaluated for application as semiconducting materials (Table 2). P4 showed the most red-shifted absorption and the highest HOMO energy level among P1–P4 because of its strong donor unit (bithiophene). The planar structure of the main chain also contributed to the extended π conjugation (Fig. S18, ESI†). The planarity and donor–acceptor structure led to the aggregation of the polymer, resulting in the lowest solubility and largest roughness of the thin film among the synthesized polymers, which was confirmed by AFM (Fig. S19, ESI†). To evaluate their p-type semiconducting properties, OFETs with these polymers were fabricated.26,38,39 The fabricated devices with P4 exhibited typical p-channel thin-film transistor characteristics with well-resolved linear and saturation regimes (Fig. S20, ESI†). Based on the transfer characteristics, the average hole mobility was calculated to be 2.2 ± 0.6 × 10−5 cm2 V−1 s−1. In contrast, OFETs with P2 and P3 showed negligible hole transporting properties because their low-lying HOMO levels led to high hole injection barriers. In terms of emission properties, P2 showed a relatively high emission quantum yield. The electroluminescence (EL) properties of P2 were also evaluated in an OLED device. The EL spectrum was similar to the photoluminescence (PL) spectrum of P2 (Fig. 3). The coordinates of the CIE chromaticity diagram were x = 0.34, y = 0.57 at 2.4 mA cm−2. The maximal brightness of EL was 1915 cd m−2 at a current of 351.2 mA cm−2, and the maximal external quantum efficiency (EQE) of the OLED was 0.31% at 5.4 mA cm−2 (Fig. S21, ESI†). These results showed that P2 served as emitting material of the OLED device.
| λ max/nm | λ em/nm | Φ /% |
E
optgb/eV |
HOMOc/eV | LUMOd/eV | |
|---|---|---|---|---|---|---|
| a Emission quantum yield. b Estimated from the absorption onset. c Estimated from photoelectron yield spectroscopy. d E LUMO = Eoptg + EHOMO. | ||||||
| P1 | 410 | 520 | 6.8 | 2.67 | −6.11 | −3.44 |
| P2 | 442 | 530 | 10.4 | 2.50 | −6.12 | −3.62 |
| P3 | 455 | 568 | 2.7 | 2.40 | −5.69 | −3.29 |
| P4 | 539 | 682 | 1.2 | 2.00 | −5.47 | −3.47 |
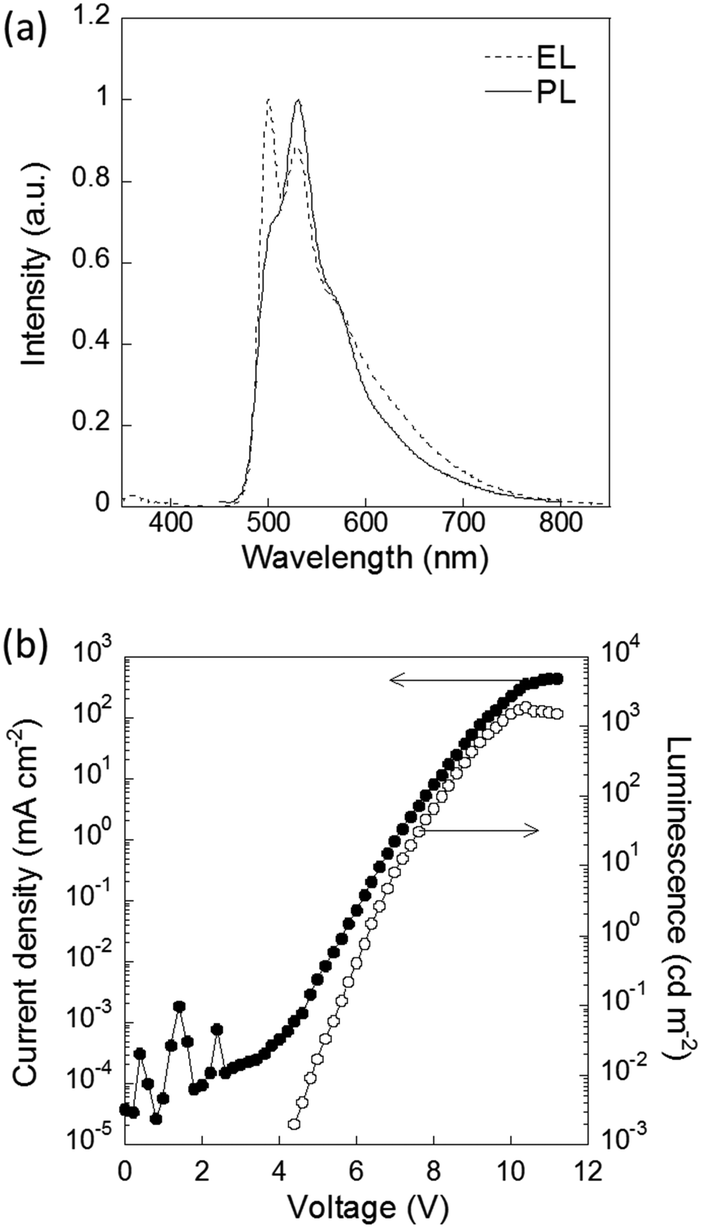 | ||
| Fig. 3 (a) PL spectrum of a thin film of P2 and the EL spectrum of an OLED with P2 at 7 V. (b) Current density and luminescence characteristics of the device. | ||
Conclusion
In this study, a new synthetic strategy for semiconducting polymers was developed using a Cu-catalysed aerobic oxidative C–H/C–H coupling reaction. This reaction proceeded with bench-stable Cu(OAc)2 as a catalyst and air as the oxidant source under neutral conditions. These simple conditions are beneficial for the green synthesis of semiconducting materials in comparison with the previously reported aerobic oxidative polymerization methods that require precious metal catalysts, a large amount of the metal co-oxidant, and harsh reaction conditions. The use of direct C–H arylation in monomer synthesis reduced the overall number of synthetic steps. Since the synthesized polymer exhibited semiconducting properties in OFETs and OLEDs, this synthetic strategy will be a reliable tool for the preparation of semiconducting materials.Experimental section
General procedure for polycondensation (Table 1, entry 4). To a 25 mL Schlenk tube, 2,7-di(thiazol-5-yl)-9,9-bis(2-ethylhexyl)fluorene (M2) (130 mg, 0.23 mmol), Cu(OAc)2 (4.2 mg, 0.023 mmol), and p-xylene (0.47 mL) were added. A drying tube with calcium chloride was attached to the Schlenk tube. The mixture was gently stirred and heated at 140 °C for 24 h under air. Volatiles were removed in vacuo and organic residues were dissolved in CHCl3. To the solution, aqueous solution of EDTA·2Na was added. After stirring for overnight, the organic phase was separated and washed with water. The organic layer was passed through Celite, and concentrated in vacuo. Reprecipitation from CHCl3/methanol and washing with hexane yielded P2 as a yellow solid (128 mg, 98%). Mn = 19800, Mw/Mn = 3.73. 1H NMR (400 MHz, CDCl3): δ 8.12 (s, 2H), 7.80 (d, 2H, J = 8.4 Hz), 7.66 (m, 4H), 2.09 (br, 2H), 0.98–0.71 (br, 16H), 0.65 (br, 6H), 0.57 (br, 8H). 13C{1H} NMR (100 MHz, CDCl3): δ 158.9, 150.9, 141.1, 140.3, 138.2, 128.4, 124.9, 121.5, 119.6, 54.2, 43.3, 33.7, 32.8, 27.1, 26.1, 21.6, 12.9, 9.34. Elemental analysis: calculated for C35H42N2S2: C, 75.76; H, 7.63; N, 5.05. Found: C, 74.49; H, 7.77; N, 4.94.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Chemical Analysis Center of University of Tsukuba for the measurements of NMR spectra, ICP-AES, and elemental analysis. The authors also thank to Prof. D. Takeuchi, Prof. Y. Nishihara, and Prof. H. Mori for elemental analysis and high temperature GPC measurements. This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas “π-System Figuration” (JP17H05141), Scientific Research (17K05973 and 17H03063).Notes and references
- A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed.
- P. M. Beaujuge and J. M. J. Fréchet, J. Am. Chem. Soc., 2011, 133, 20009–20029 CrossRef CAS PubMed.
- M. Gsänger, D. Bialas, L. Huang, M. Stolte and F. Würthner, Adv. Mater., 2016, 28, 3615–3645 CrossRef PubMed.
- C. Liu, K. Wang, X. Gong and A. J. Heeger, Chem. Soc. Rev., 2016, 45, 4825–4846 RSC.
- V. Vohra, K. Kawashima, T. Kakara, T. Koganezawa, I. Osaka, K. Takimiya and H. Murata, Nat. Photonics, 2015, 9, 403–408 CrossRef CAS.
- R. Po, A. Bernardi, A. Calabrese, C. Carbonera, G. Corso and A. Pellegrino, Energy Environ. Sci., 2014, 7, 925–943 CAS.
- K. Okamoto, J. Zhang, J. B. Housekeeper, S. R. Marder and C. K. Luscombe, Macromolecules, 2013, 46, 8059–8078 CrossRef CAS.
- J.-R. Pouliot, F. Grenier, J. T. Blaskovits, S. Beaupré and M. Leclerc, Chem. Rev., 2016, 116, 14225–14274 CrossRef CAS PubMed.
- Q. Wang, R. Takita, Y. Kikuzaki and F. Ozawa, J. Am. Chem. Soc., 2010, 132, 11420–11421 CrossRef CAS PubMed.
- W. Lu, J. Kuwabara and T. Kanbara, Macromolecules, 2011, 44, 1252–1255 CrossRef CAS.
- L. A. Estrada, J. J. Deininger, G. D. Kamenov and J. R. Reynolds, ACS Macro Lett., 2013, 2, 869–873 CrossRef CAS.
- J. Kuwabara, T. Yasuda, S. J. Choi, W. Lu, K. Yamazaki, S. Kagaya, L. Han and T. Kanbara, Adv. Funct. Mater., 2014, 24, 3226–3233 CrossRef CAS.
- Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787–8863 CrossRef CAS PubMed.
- J. K. Park, J. Jo, J. H. Seo, J. S. Moon, Y. D. Park, K. Lee, A. J. Heeger and G. C. Bazan, Adv. Mater., 2011, 23, 2430–2435 CrossRef CAS PubMed.
- J. Kuwabara, T. Yasuda, N. Takase and T. Kanbara, ACS Appl. Mater. Interfaces, 2016, 8, 1752–1758 CAS.
- D. Son, T. Kuwabara, K. Takahashi and K. Marumoto, Appl. Phys. Lett., 2016, 109, 133301 CrossRef.
- N. Toshima and S. Hara, Prog. Polym. Sci., 1995, 20, 155–183 CrossRef CAS.
- N. Toshima, K. Kanaka, A. Koshirai and H. Hirai, Bull. Chem. Soc. Jpn., 1988, 61, 2551–2557 CrossRef CAS.
- Q. Huang, X. Qin, B. Li, J. Lan, Q. Guo and J. You, Chem. Commun., 2014, 50, 13739–13741 RSC.
- S. Habaue, T. Seko and Y. Okamoto, Macromolecules, 2002, 35, 2437–2439 CrossRef CAS.
- Q. Guo, D. Wu and J. You, ChemSusChem, 2016, 9, 2765–2768 CrossRef CAS PubMed.
- K. Tsuchiya and K. Ogino, Polym. J., 2013, 45, 281–286 CrossRef CAS.
- L. Ricciotti, F. Borbone, A. Carella, R. Centore, A. Roviello, M. Barra, G. Roviello, C. Ferone, C. Minarini and P. Morvillo, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4351–4360 CrossRef CAS.
- Y. Lin, H. Fan, Y. Li and X. Zhan, Adv. Mater., 2012, 24, 3087–3106 CrossRef CAS PubMed.
- H. L. Su, D. N. Sredojevic, H. Bronstein, T. J. Marks, B. C. Schroeder and M. Al-Hashimi, Macromol. Rapid Commun., 2017, 38, 1600610 CrossRef PubMed.
- D. H. Kim, B. L. Lee, H. Moon, H. M. Kang, E. J. Jeong, J. Il Park, K. M. Han, S. Lee, B. W. Yoo, B. W. Koo, J. Y. Kim, W. H. Lee, K. Cho, H. A. Becerril and Z. Bao, J. Am. Chem. Soc., 2009, 131, 6124–6132 CrossRef CAS PubMed.
- B. Balan, C. Vijayakumar, A. Saeki, Y. Koizumi and S. Seki, Macromolecules, 2012, 45, 2709–2719 CrossRef CAS.
- D. Monguchi, A. Yamamura, T. Fujiwara, T. Somete and A. Mori, Tetrahedron Lett., 2010, 51, 850–852 CrossRef CAS.
- Y. Li, J. Jin, W. Qian and W. Bao, Org. Biomol. Chem., 2010, 8, 326–330 CAS.
- M. Zhu, K. Fujita and R. Yamaguchi, Chem. Commun., 2011, 47, 12876–12878 RSC.
- B. Liégault, D. Lapointe, L. Caron, A. Vlassova and K. Fagnou, J. Org. Chem., 2009, 74, 1826–1834 CrossRef PubMed.
- J. Kuwabara, M. Sakai, Q. Zhang and T. Kanbara, Org. Chem. Front., 2015, 2, 520–525 RSC.
- Q. Guo, R. Jiang, D. Wu and J. You, Macromol. Rapid Commun., 2016, 37, 794–798 CrossRef CAS PubMed.
- M. Kuramochi, J. Kuwabara, W. Lu and T. Kanbara, Macromolecules, 2014, 47, 7378–7385 CrossRef CAS.
- S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234–6458 CrossRef CAS PubMed.
- S. D. McCann and S. S. Stahl, Acc. Chem. Res., 2015, 48, 1756–1766 CrossRef CAS PubMed.
- M. A. Dubé and S. Salehpour, Macromol. React. Eng., 2014, 8, 7–28 CrossRef.
- H. Nagashima, M. Saito, H. Nakamura, T. Yasuda and T. Tsutsui, Org. Electron., 2010, 11, 658–663 CrossRef CAS.
- D. Chen, Y. Zhao, C. Zhong, G. Yu, Y. Liu and J. Qin, Sci. China: Chem., 2012, 55, 760–765 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: For experimental details, NMR, UV-vis absorption and photoemission spectra. See DOI: 10.1039/c7qm00584a |
| This journal is © the Partner Organisations 2018 |