DOI:
10.1039/C7QM00582B
(Research Article)
Mater. Chem. Front., 2018,
2, 807-814
Highly efficient transformation of linear poly(phenylene ethynylene)s into zigzag-shaped π-conjugated microporous polymers through boron-mediated alkyne benzannulation†
Received
14th December 2017
, Accepted 6th February 2018
First published on 7th February 2018
Abstract
Porous polymers offer great advantages compared to other microporous materials in terms of solubility and processability. However, the design of porous polymers has suffered from the limited availability of suitable building blocks. Here we propose a conceptually new strategy for the design of porous polymers, which involves the transformation of a rigid linear polymer into a rigid zigzag polymer with a large free volume around the polymer backbone. This strategy relies on a boron-mediated alkyne benzannulation reaction, which was recently developed by our group. When the benzannulation reaction was applied to poly(phenylene ethynylene) (PPE) derivatives, a linear-to-zigzag structural transformation successfully occurred to give the corresponding π-conjugated polymers with a diarylphenanthrene unit in the main chain. As revealed by N2 adsorption experiments, while the parent PPEs were non-porous, the zigzag polymers in the solid state possessed porosity with a specific surface area of up to 366 m2 g−1, where the surface area largely depended on the steric bulkiness of the substituents on the polymer. Considering the fact that a wide variety of PPE derivatives have so far been synthesized, the present strategy may open a new avenue for the development of functional porous polymers.
Introduction
Microporous materials have attracted considerable attention because they have a wide variety of applications, including gas storage and separation, catalysis, ion transport and drug delivery.1 As represented by metal–organic frameworks (MOFs),1a–d,2 covalent-organic frameworks (COFs)1e,3 and highly crosslinked porous π-conjugated polymers,4 many porous materials have been developed, which have three-dimensional robust network structures formed by metal ion-to-ligand coordination and/or covalent linkages to give a particular space in the solid state. However, these porous materials suffer from insolubility and low processability due to their highly crosslinked network structures. In this regard, non-crosslinked porous polymers offer the advantage of high solubility, which is beneficial for film fabrication and thus enables membrane-based applications.
A key for the design of porous polymers is how to make free volume around the polymer backbone in the solid state.5,6 As represented by poly[1-(trimethylsilyl)-1-propyne], rigid polyacetylenes carrying sterically bulky substituents are known to provide a microporous solid.5 Recently, McKeown and co-workers have reported an interesting class of microporous polymers, called polymers of intrinsic microporosity (PIM).1f,g,6 PIMs are characterized by their rigid and randomly contorted ladder-type backbones, which provide high solubility as well as processability, to allow the fabrication of free-standing microporous membranes.6 However, the limited availability of suitable building blocks for the construction of soluble microporous polymers might be an obstacle for the further development of polymer-based porous materials with various functionalities such as redox, catalytic and sensing activities.5,6 Therefore, a new design concept of porous polymers, which realizes both design flexibility and processability, is highly desired.
We conceived a new design strategy for the construction of porous polymers based on the structural transformation of a rigid linear polymer into a rigid zigzag-shaped polymer (Fig. 1a). As expected from the structural feature of PIMs, zigzag polymers with high rigidity may offer a large free volume around the polymer backbone, thereby leading to a porous solid upon aggregation (Fig. 1a). To embody this strategy, we used a recently developed benzannulation reaction of alkyne derivatives with 9-chloro-9-borafluorene7 (1H), which enables the facile and efficient transformation of various diarylacetylenes into the corresponding 9,10-diarylphenanthrenes by a one-pot reaction protocol (Fig. 1b).8 The first step of this reaction is 1,2-carboboration of diarylacetylene with 1H to form borepin, which has a boron-containing seven-membered ring (Fig. 1b). Upon one-electron oxidation of the resulting borepin, deborylation/C–C coupling takes place to afford 9,10-diarylphenanthrene (Fig. 1b). From a geometrical point of view, a linear alkyne moiety is bent by 120 degrees through the benzannulation reaction (Fig. 1). Here we report that the boron-mediated benzannulation can be successfully applied to linear poly(phenylene ethynylene)s (PPEs)9 to give π-conjugated zigzag polymers (Fig. 1 and Scheme 1). N2 adsorption–desorption experiments revealed that the obtained diarylphenanthrene-based zigzag polymers in the solid state possess a microporous structure with a Brunauer–Emmett–Teller (BET) surface area (SBET) of up to 366 m2 g−1, where the sterically bulky substituents on the diarylphenanthrene are essential for achieving microporosity. Considering the high design flexibility of PPEs as well as the wide substrate scope of the boron-mediated benzannulation,8 the present design strategy is promising for the development of porous polymers with various functionalities.
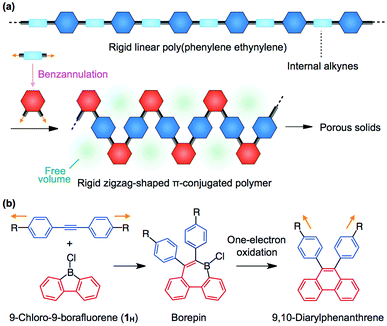 |
| | Fig. 1 (a) Schematic illustration of the design strategy for constructing a porous polymer through the structural transformation of linear PPE into a zigzag-shaped diarylphenanthrene-based polymer by benzannulation of the internal alkyne units in the main chain of PPE. (b) Schematic illustration of a borafluorene-mediated alkyne benzannulation reaction that allows the facile transformation of diarylacetylene into 9,10-diarylphenanthrene. Orange arrows indicate the direction of chemical bonding. | |
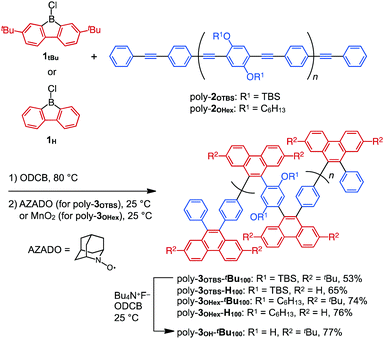 |
| | Scheme 1 Synthesis of zigzag-shaped π-conjugated poly-3 (fully benzannulated) by the boron-mediated benzannulation of poly(phenylene ethynylene) poly-2 with borafluorene 1. TBS = tert-butyldimethylsilyl and ODCB = o-dichlorobenzene. | |
Results and discussion
Synthesis of rigid zigzag-shaped π-conjugated polymers with a diarylphenanthrene backbone
As precursors for zigzag polymers, we used 2,7-di-tert-butyl-9-chloro-9-borafluorene (1tBu) in addition to non-substituted 1H (Scheme 1). We also chose PPE with bulky tert-butyldimethylsilyl (TBS)-protected phenol groups on the phenylene rings (poly-2OTBS, Mn = 6400, PDI = 2.02) as well as PPE having flexible hexyloxy side chains (poly-2OHex, Mn = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400, PDI = 4.50) (Scheme 1).10 The benzannulation reactions of PPEs with these borafluorenes afforded four types of zigzag polymers, each with a different steric environment (Scheme 1).
400, PDI = 4.50) (Scheme 1).10 The benzannulation reactions of PPEs with these borafluorenes afforded four types of zigzag polymers, each with a different steric environment (Scheme 1).
Typically, poly-2OTBS was reacted with 1tBu in o-dichlorobenzene (ODCB) at 80 °C, and the resulting mixture was treated with 2-azaadamantane-N-oxyl (AZADO) as a one-electron oxidant. By reprecipitation of the reaction mixture in MeOH, fully benzannulated poly-3OTBS-tBu100 was obtained as an orange powder in 53% yield. As shown in Fig. 2, the FT-IR spectrum of poly-3OTBS-tBu100 was devoid of a C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C stretching band at 2205 cm−1, which was observed for poly-2OTBS. The 1H NMR spectrum (500 MHz) of poly-3OTBS-tBu100 in CDCl3 at 25 °C displayed broad signals in the aromatic region at δ = 8.61 and 8–6 ppm, which coincide with the 1H NMR signals of 9,10-diarylphenanthrene derivatives.8 Thus, the alkyne units of PPE were successfully benzannulated (Fig. 3). The electronic absorption spectrum of poly-3OTBS-tBu100 in CH2Cl2 at 25 °C (Fig. S1, ESI†) showed that the absorption maximum of poly-3OTBS-tBu100 (λmax = 264 nm) was significantly blue-shifted relative to that of parent poly-2OTBS (λmax = 389 nm), suggesting that the effective π-conjugation length of poly-3OTBS-tBu100 is shorter than that of poly-2OTBS due to the zigzag-shaped structure. Interestingly, in the gel permeation chromatography (GPC) analysis, the retention time of benzannulated poly-3OTBS-tBu100 was longer than that of poly-2OTBS (Fig. S2, ESI†), suggesting that the hydrodynamic volume of the polymer decreases upon bending.11 In addition to fully benzannulated poly-3OTBS-tBu100, we also prepared poly-3OTBS-tBu50 (Fig. 2, 3, Fig. S15, S16 and Scheme S1, ESI†), in which half of the alkyne units are benzannulated (Fig. 2), so that we could evaluate the effect of the benzannulation ratio on the solid-state properties. In a manner similar to that for poly-3OTBS-tBu100, fully benzannulated poly-3OTBS-H100, poly-3OHex-tBu100 and poly-3OHex-H100 were obtained as orange powders in respective yields of 65, 74 and 76%, and unambiguously characterized by FT-IR spectroscopy as well as 1H and 13C NMR spectroscopy (Fig. S17–S21, ESI†). The obtained zigzag polymers were sufficiently soluble in common organic solvents such as chloroform, dichloromethane and toluene and thus allowed for not only the full structural characterization but also solution processing of the material.
C stretching band at 2205 cm−1, which was observed for poly-2OTBS. The 1H NMR spectrum (500 MHz) of poly-3OTBS-tBu100 in CDCl3 at 25 °C displayed broad signals in the aromatic region at δ = 8.61 and 8–6 ppm, which coincide with the 1H NMR signals of 9,10-diarylphenanthrene derivatives.8 Thus, the alkyne units of PPE were successfully benzannulated (Fig. 3). The electronic absorption spectrum of poly-3OTBS-tBu100 in CH2Cl2 at 25 °C (Fig. S1, ESI†) showed that the absorption maximum of poly-3OTBS-tBu100 (λmax = 264 nm) was significantly blue-shifted relative to that of parent poly-2OTBS (λmax = 389 nm), suggesting that the effective π-conjugation length of poly-3OTBS-tBu100 is shorter than that of poly-2OTBS due to the zigzag-shaped structure. Interestingly, in the gel permeation chromatography (GPC) analysis, the retention time of benzannulated poly-3OTBS-tBu100 was longer than that of poly-2OTBS (Fig. S2, ESI†), suggesting that the hydrodynamic volume of the polymer decreases upon bending.11 In addition to fully benzannulated poly-3OTBS-tBu100, we also prepared poly-3OTBS-tBu50 (Fig. 2, 3, Fig. S15, S16 and Scheme S1, ESI†), in which half of the alkyne units are benzannulated (Fig. 2), so that we could evaluate the effect of the benzannulation ratio on the solid-state properties. In a manner similar to that for poly-3OTBS-tBu100, fully benzannulated poly-3OTBS-H100, poly-3OHex-tBu100 and poly-3OHex-H100 were obtained as orange powders in respective yields of 65, 74 and 76%, and unambiguously characterized by FT-IR spectroscopy as well as 1H and 13C NMR spectroscopy (Fig. S17–S21, ESI†). The obtained zigzag polymers were sufficiently soluble in common organic solvents such as chloroform, dichloromethane and toluene and thus allowed for not only the full structural characterization but also solution processing of the material.
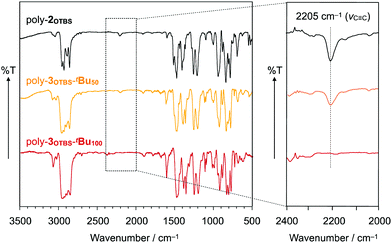 |
| | Fig. 2 IR spectra (KBr) and magnification of poly-3OTBS-tBu100, poly-3OTBS-tBu50 and poly-2OTBS at 25 °C. The area ratio of the peak due to the CC stretching band (2205 cm−1) is 2.1:1.0 (poly-2OTBS:poly-3OTBS-tBu50). | |
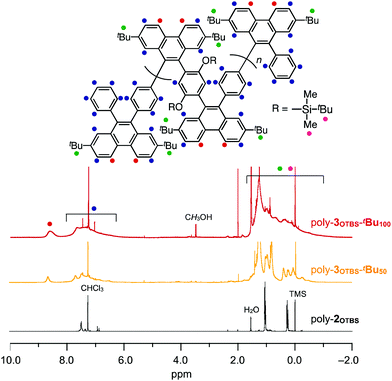 |
| | Fig. 3
1H NMR spectra (500 MHz) of poly-3OTBS-tBu100 (red), poly-3OTBS-tBu50 (orange) and poly-2OTBS (black) in CDCl3 at 25 °C. TMS = tetramethylsilane. | |
Substituent effect on the porosity of diarylphenanthrene-based zigzag π-conjugated polymers
To evaluate the porosity of the diarylphenanthrene-based zigzag polymers, we conducted N2 adsorption experiments. Fig. 4 shows the N2 adsorption–desorption isotherms obtained for powder samples of poly-3OTBS-tBu100, poly-3OTBS-tBu50, poly-3OTBS-H100 and poly-2OTBS at 77 K. As expected, poly-3OTBS-tBu100 displayed a hysteretic N2 adsorption–desorption curve (Fig. 4a), the shape of which can be classified as type I according to the IUPAC classification.12 The SBET value of poly-3OTBS-tBu100 was estimated to be 366 m2 g−1. Based on the Barrett–Joyner–Halenda (BJH) analysis (Fig. S2, ESI†), the pore size of poly-3OTBS-tBu100 was considered to be approximately 2 nm. While the observed SBET value is not very high compared to those of other microporous polymers,5,6poly-3OTBS-tBu100 surely possesses micropores in the solid state. Note that since the parent linear poly-2OTBS is nonporous (Fig. 4d), the microporosity of poly-3OTBS-tBu100 should arise from its rigid zigzag-shaped structure, which is brought about by the benzannulation of the linear poly(phenylene ethynylene) backbone (Fig. 1).
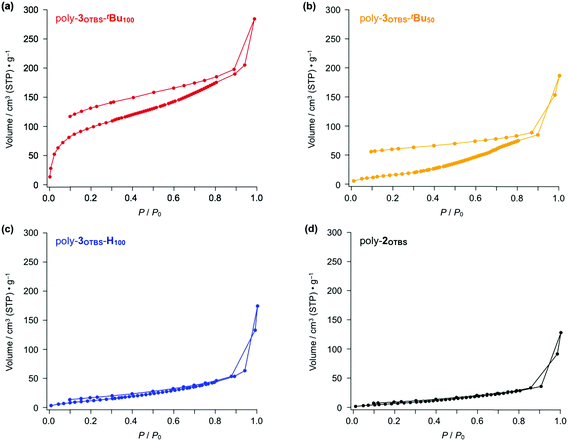 |
| | Fig. 4 N2 adsorption–desorption isotherms of powder samples of (a) poly-3OTBS-tBu100, (b) poly-3OTBS-tBu50, (c) poly-3OTBS-H100 and (d) parent poly-2OTBS (Mn = 6400, PDI = 2.02) at 77 K. | |
The benzannulation ratio of the polymer backbone greatly affected the porosity of the zigzag polymer. The N2 adsorption–desorption isotherm showed that a powder sample of poly-3OTBS-tBu50 (Scheme S1, ESI†), in which half of the alkyne units are benzannulated, hardly possessed micropores (Fig. 4b). The estimated SBET value for poly-3OTBS-tBu50 (64.9 m2 g−1) was significantly smaller than that of poly-3OTBS-tBu100 (366 m2 g−1), and the BJH analysis suggested that the pore-size distribution for poly-3OTBS-tBu50, centered at 6.4 nm, was much larger than that for poly-3OTBS-tBu100 (Fig. S4, ESI†). Thus, formation of a microporous structure is hampered for poly-3OTBS-tBu50, most likely because alkyne and benzannulated units are randomly distributed in the polymer backbone (Scheme S1, ESI†).
Since poly-3OTBS-H100, which lacks tBu groups at the phenanthrene units, was hardly microporous (Fig. 4c, blue), the sterically bulky tBu groups are considered to be essential for endowing the microporous structure with poly-3OTBS-tBu100. Even in the presence of tBu groups at the phenanthrene units, the zigzag polymer loses porosity, when the bulky TBS groups are replaced by flexible hexyl groups (poly-3OHex-tBu100, Fig. 5, red). As expected, poly-3OHex-H100 was also found to be nonporous (Fig. 5, blue).
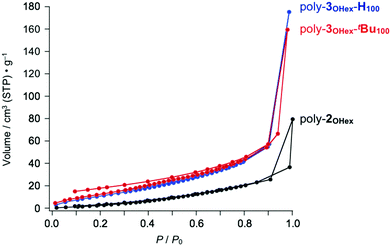 |
| | Fig. 5 N2 adsorption isotherms of powder samples of poly-3OHex-tBu100 (red), poly-3OHex-H100 (blue) and parent poly-2OHex (black, Mn = 10400, PDI = 4.50) at 77 K. | |
Deprotection of the TBS groups of a zigzag polymer (poly-3OTBS-tBu100)
According to the N2 adsorption–desorption isotherm profiles in Fig. 4 and 5, we initially thought that both bulky tBu and TBS groups are required to ensure the porosity of the zigzag polymers. Unexpectedly, however, removal of the TBS groups of poly-3OTBS-tBu100 did not significantly affect the microporosity. For example, an ODCB solution of poly-3OTBS-tBu100, synthesized from poly-2OTBS (Mn = 5900, PDI = 2.32), was treated with tetrabutylammonium fluoride (Bu4N+F−) at 25 °C, affording poly-3OH-tBu100 as an orange powder in 77% yield (Scheme 1). FT-IR spectroscopy and 1H and 13C NMR spectroscopy of poly-3OH-tBu100 confirmed the absence of TBS groups (Fig. S5, S22 and S23, ESI†). As shown in Fig. 6, the N2 adsorption–desorption isotherm of a powder sample of poly-3OH-tBu100 at 77 K (green) was very similar to that of the precursor poly-3OTBS-tBu100 under identical measurement conditions (red). The SBET and BJH pore size for poly-3OH-tBu100 were estimated to be 283.9 m2 g−1 and ∼2 nm, respectively, which were comparable to those obtained for poly-3OTBS-tBu100 (SBET: 303.9 m2 g−1, BJH pore size: 2.4 nm). Therefore, even after the removal of the large TBS groups, the free volume around the diarylphenanthrene backbone is not impaired, to preserve microporosity in the solid state. The fact that the diarylphenanthrene-based zigzag π-conjugated polymer allows chemical transformation while maintaining microporosity is noteworthy.
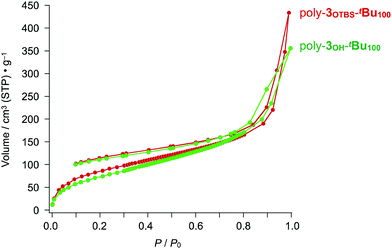 |
| | Fig. 6 N2 adsorption isotherms at 77 K of powder samples of poly-3OTBS-tBu100 (red) and poly-3OH-tBu100 (green), both of which were converted from poly-2OTBS (Mn = 5900, PDI = 2.31). | |
Structural requirements for microporosity in diarylphenanthrene-based zigzag polymers
Table 1 summarizes the results of the N2 adsorption experiments in this study. As mentioned above, the zigzag polymers have micropores with a size of approximately 2 nm. Obviously, not only the rigid zigzag-shaped backbone but also sterically bulky tBu groups on the phenanthrene units are needed to achieve microporosity (entries 1 and 3). The comparison between entries 1 and 6 indicates that the molecular weight and PDI may affect the porosity to some extent. Meanwhile, the microporosity of the zigzag polymers is impaired when the alkyne units of PPEs are insufficiently benzannulated (entries 1 and 2). Flexible chains such as a hexyloxy group may also hamper the formation of micropores, most likely through filling voids (entries 4 and 5). If the above-listed structural requirements are satisfied, further chemical modification of the zigzag polymers may become possible while maintaining porosity (entries 6 and 7).
Table 1 Specific surface areas (SBET) and BJH pore sizes of poly-3
| Entry |
Polymer |
S
BET (m2 g−1) |
Pore size (nm) |
|
Synthesized from poly-2OTBS (Mn = 6400, PDI = 2.02).
Synthesized from poly-2OTBS (Mn = 5900, PDI = 2.32).
|
| 1 |
Poly-3OTBS-tBu100
|
365.5 |
∼2 |
| 2 |
Poly-3OTBS-tBu50
|
64.9 |
6.4 |
| 3 |
Poly-3OTBS-H100
|
Nonporous |
— |
| 4 |
Poly-3OHex-tBu100
|
Nonporous |
— |
| 5 |
Poly-3OHex-H100
|
Nonporous |
— |
| 6 |
Poly-3OTBS-tBu100
|
303.9 |
2.4 |
| 7 |
Poly-3OH-tBu100
|
283.9 |
∼2 |
Conclusions
We have demonstrated the successful synthesis of a new type of π-conjugated zigzag polymer by the benzannulation reaction of PPEs with borafluorene derivatives. Importantly, the boron-mediated benzannulation8 was highly effective even for polymeric substances with multiple reaction sites, thus enabling quantitative conversion of the internal alkynes in the main chain of PPEs into diarylphenanthrene units. The resulting polymers possess a large free volume around the main chain to exhibit microporosity. Basically, the formation of the microporous structure in the rigid zigzag-shaped polymers can be rationalized in light of the general guideline for the design of organic and polymeric microporous materials.13 However, the present system is remarkable in that it utilizes the structural transformation of inherently nonporous linear PPEs, and the π-conjugated systems can be maintained after the polymer reaction. Furthermore, owing to the wide substrate scope of the benzannulation reaction,8 various functional PPEs can be used as precursors for constructing zigzag polymers,9 which would be beneficial for the design of microporous materials with synergistic properties (e.g., redox, catalytic and sensing activities) in combination with gas adsorption/permeation activity. In this context, poly-3OH-tBu100 with a redox active hydroquinone backbone may be interesting. Taking all the above features into account, the present design strategy for porous, zigzag, and π-conjugated polymers may open a new avenue for the development of functional porous polymeric materials. Further studies along these lines are now underway.
Experimental section
Materials
Air- and/or moisture-sensitive compounds were handled in a glove box under argon. Unless otherwise noted, all commercial reagents were used as received. o-Dichlorobenzene (ODCB) was dried over CaH2 and freshly distilled prior to use. 9-Chloro-9-borafluorene (1H) was prepared according to a previously reported procedure.7
Methods
Analytical GPC was performed at 40 °C regulated using a column oven on a TOSOH GPC-8020 system equipped with a refraction index (RI) detector and a UV detector (UV-8020), using chloroform (CHCl3) as an eluent at a flow rate of 0.40 mL min−1 on a column (Shodex LF-804). The molecular weight calibration curve was obtained by using standard polystyrenes (TSKstandard polystyrene, TOSOH). Size-exclusion chromatography–multi-angle laser light scattering (SEC–MALS) measurements were performed on a TOSOH GPC-8020 system, equipped with a Shodex KF-805L column, a Wyatt Technology DAWN HELEOS multi-angle laser light scattering photometer with a He–Ne laser (λ0 = 658 nm) and a Wyatt Technology Optilab rEX differential refractometer, using chloroform (CHCl3) as an eluent at a flow rate of 0.40 mL min−1. The SEC-MALS profiles were recorded and analyzed using ASTRA software (ver.6.1.1, Wyatt Technology). Differential refractive index increments (∂n/∂c) for the polymers in CHCl3 were determined based on the peak area of the refractive index chromatograms and the polymer mass concentration (c) of injected solutions assuming that all the injected polymers were fully recovered. The ∂n/∂c values of poly-3OTBS-tBu100, poly-3OTBS-tBu50, poly-3OTBS-H100, poly-3OHex-tBu100, poly-3OHex-H100 and poly-3OH-tBu100 were determined to be 0.110, 0.152, 0.135, 0.138, 0.164 and 0.072 cm3 g−1, respectively, at λ0 = 658 nm. Angular dependences of the scattering intensity were analyzed using the Debye plot in a scattering angle range from 45.8° to 138.8° to estimate the weight-averaged molecular weight Mw and the z-averaged mean-square radius of gyration 〈S2〉z of each fraction. Infrared (IR) spectra were recorded at 25 °C on a JASCO FT/IR-660Plus Fourier-transform infrared spectrometer. NMR spectroscopy measurements were carried out at 25 °C on a Bruker AVANCE-500 spectrometer (500 MHz for 1H, 125 MHz for 13C and 128 MHz for 11B). Chemical shifts (δ) are expressed relative to the resonances of the residual non-deuterated solvent for 1H [CDCl3: 1H(δ) = 7.26 ppm] and 13C [CDCl3: 13C(δ) = 78.16 ppm] and external BF3OEt2 in CDCl3 for 11B (11B(δ) = 0.0 ppm). Absolute values of the coupling constants are given in Hertz (Hz), regardless of their sign. Multiplicities are abbreviated as singlet (s), doublet (d), doublet of doublets (dd), triplet (t), quartet (q), multiplet (m), and broad (br). N2 adsorption measurements were performed at 77 K after pretreatment at 150 °C under a reduced pressure for 3 h on a BEL Japan Belsorp-mini II apparatus. The Brunauer–Emmett–Teller (BET) surface areas were estimated over a relative pressure (P/P0) range of 0.05–0.20. The pore size distribution was obtained from the analysis of the adsorption branch of the isotherms using the BJH (Barrett–Joyner–Halenda) method.
Synthesis
2,7-Di-tert-butyl-9-chloro-9-borafluorene (1tBu).
Under argon at 25 °C, a hexane solution (2.69 M) of n-BuLi (17.5 mL, 4.45 mmol) was added dropwise to a dry benzene solution (20 mL) of 2,2′-dibromo-4,4′-di-tert-butyl-1,1′-biphenyl14 (7.00 g, 165 mmol), and the resulting mixture was stirred at 80 °C for 4 days, forming a white suspension. After the reaction mixture was allowed to cool to 25 °C, a hexane solution (1.0 M) of BCl3 (17.3 mL, 173 mmol) was slowly added to the resulting suspension, where the color of the reaction mixture turned from white to yellow. The reaction mixture was stirred at 25 °C for 1 day and evaporated under reduced pressure. To the residue was added dry hexane (100 mL), and the resulting suspension was filtered through a glass filter. The filtrate was collected, evaporated under reduced pressure and recrystallized to give 1tBu (2.47 g, 18.7 mmol) as a yellow powder in 48% yield: 1H NMR (500 MHz, CDCl3, 25 °C): δ (ppm) 7.56 (s, 2H), 7.35 (d, J = 8.0 Hz, 2H), 7.25–7.18 (m, 2H), 1.32 (s, 18H). 11B NMR (128 MHz, CDCl3, 25 °C): δ (ppm) 64.2. 13C NMR (125 MHz, CDCl3, 25 °C): δ (ppm) 151.2, 150.5, 138.7 (br), 131.9, 129.8, 119.2, 34.7, 31.2. 1H, 11B and 13C NMR spectra of 1tBu are shown in Fig. S4, S5 and S6 (ESI†), respectively.
Poly-2OTBS
.
Poly-2OTBS was synthesized according to a slightly modified procedure reported by Lei et al.10b Under argon, bis(triphenylphosphine)palladium(II) dichloride (59.4 mg, 0.0845 mmol) was added to a dry toluene suspension (50 mL) of a mixture of 1,4-bis(tert-butyldimethylsilyloxyl)-2,5-diiodobenzene15 (1.00 g, 1.69 mmol), 1,4-diethynylbenzene (214 mg, 1.69 mmol), triphenylphosphine (66.6 mg, 0.254 mmol), copper(I) iodide (32.3 mg, 0.170 mmol) and dry trimethylamine (3.53 mL, 25.4 mmol), and the reaction mixture was stirred at 80 °C for 3 h. To the mixture was added iodobenzene (0.0346 g, 0.170 mmol), and the resulting mixture was stirred at 80 °C for 1 h, allowed to cool to 25 °C, poured into water (50 mL) and extracted with toluene. The organic layer was washed with water, partially evaporated under reduced pressure until the volume of the solution was reduced to ∼25 mL. The resulting concentrated solution was poured into vigorously stirred MeOH (300 mL). The resulting orange precipitate was collected by filtration, washed with MeOH (10 mL × 3 times) and dried under reduced pressure. Repeated reprecipitation of the resultant solid, using toluene (good solvent) and MeOH (poor solvent), afforded poly-2OTBS (0.749 g) as a yellow powder in 83% yield: FT-IR (KBr): ν (cm−1) 3075, 3047, 2952, 2205 (vCC), 1906, 1792, 1684, 1599, 1515, 1472, 1434, 1402, 1362, 1277, 1255, 1207, 1096, 999, 936, 881, 838, 803, 781, 754, 743, 691, 626, 541, 519. 1H NMR (500 MHz, CDCl3, 25 °C): δ (ppm) 7.49 (br, 6H), 1.06–1.03 (m, 18H), 0.29–0.24 (m, 12H). 13C NMR (125 MHz, CDCl3, 25 °C): δ (ppm) 150.9, 150.5, 150.4, 149.6, 135.2, 134.3, 132.6, 131.6, 130.4, 129.8, 129.2, 128.5, 128.4, 127.9, 124.5, 123.8, 123.3, 121.4, 116.9, 116.1, 94.2, 91.4, 88.8, 82.3, 25.9, 18.5, 18.4, −3.9, −4.1. Mn = 6400; Mw = 12900; PDI = 2.02. Similarly, poly-2OTBS (Mn = 5900, Mw = 13600, PDI = 2.32) was obtained as an orange powder in 80% yield. 1H and 13C NMR spectra of poly-2OTBS are shown in Fig. S9 and S10 (ESI†), respectively.
Poly-2OHex
.
Poly-2OHex was synthesized according to a slightly modified procedure reported by Lei et al.10b Under argon, bis(triphenylphosphine)palladium(II) dichloride (351 mg, 0.500 mmol) was added to a dry toluene suspension (200 mL) of a mixture of 1,4-bis(hexyloxy)-2,5-diiodobenzene3 (5.00 g, 9.43 mmol), 1,4-diethynylbenzene (1.19 g, 9.43 mmol), triphenylphosphine (371 mg, 1.41 mmol), copper(I) iodide (180 mg, 0.943 mmol) and dry trimethylamine (12.0 mL, 86.1 mmol), and the reaction mixture was stirred at 80 °C for 3 h. To the mixture was added iodobenzene (1.92 g, 9.43 mmol), and the resulting mixture was stirred at 80 °C for 1 h, allowed to cool to 25 °C, poured into water (200 mL) and extracted with toluene. The organic layer was washed with water, partially evaporated under reduced pressure until the volume of the solution was reduced to ∼100 mL. The resulting concentrated solution was poured into vigorously stirred MeOH (1.0 L). The resulting orange precipitate was collected by filtration, washed with MeOH (50 mL × 3 times) and dried under reduced pressure. Repeated reprecipitation of the resultant solid, using toluene (good solvent) and MeOH (poor solvent), afforded poly-2Hex as a yellow powder (4.05 g) in 81% yield: FT-IR (KBr): ν (cm−1) 3076, 3038, 2952, 2931, 2869, 2859, 2204 (vCC), 1905, 1789, 1664, 1597, 1516, 1595, 1464, 1415, 1380, 1280, 1215, 1122, 1098, 1017, 943, 886, 857, 835, 741, 720, 692, 542, 520, 510, 489. 1H NMR (500 MHz, CDCl3, 25 °C): δ (ppm) 7.51 (br, 4H), 7.35 (br, 2H), 4.10 (t, J = 6.0 Hz, 4H), 1.85 (br, 4H), 1.37 (br, 8H), 0.91 (t, J = 6.6 Hz, 6H). 13C NMR (125 MHz, CDCl3, 25 °C): δ (ppm) 153.8, 135.0, 134.3, 132.6, 132.2, 131.8, 130.0, 129.2, 128.5, 128.4, 127.7, 124.0, 123.4, 116.1, 114.1, 69.8, 31.8, 31.7, 31.6, 29.5, 29.4, 29.3, 25.9, 25.8, 22.8, 22.7, 14.2. Mn = 10400; Mw = 46600; PDI = 4.50. 1H and 13C NMR spectra of poly-2Hex are shown in Fig. S11 and S12 (ESI†), respectively.
General procedure A: benzannulation of poly-2OTBS
Under argon, an ODCB solution (2.2 mL) of a mixture of poly-2OTBS (100 mg) and borafluorene (1tBu or 1H) was stirred at 80 °C for 72 h and allowed to cool to 25 °C. After the addition of 2-azaadamantane-N-oxyl (AZADO), the resulting mixture was stirred at 25 °C for 30 min under argon and then poured into vigorously stirred MeOH (150 mL). The resulting orange precipitate was collected by filtration, washed with MeOH (10 mL × 3 times) and dried under reduced pressure. Repeated reprecipitation of the resultant solid, using CHCl3 (good solvent) and MeOH (poor solvent), afforded a benzannulated polymer.
General procedure B: benzannulation of poly-2OHex
Under argon, an ODCB solution (2.5 mL) of a mixture of poly-2OHex (100 mg, Mn = 6400, PDI = 2.02) and borafluorene (1tBu or 1H) was stirred at 80 °C for 20 h and allowed to cool to 25 °C. After the addition of MnO2 under argon, the resulting suspension was stirred at 80 °C for 1 h and then allowed to cool to 25 °C. The resulting orange gel was dissolved by the addition of THF (5.0 mL), and insoluble materials in the resulting suspension were removed by centrifugation (15000 rpm, 15 min). The supernatant solution was collected and poured into vigorously stirred MeOH (150 mL). The resulting orange precipitate was collected by filtration, washed with MeOH (10 mL × 3 times) and dried under reduced pressure. Repeated reprecipitation of the resultant solid, using CHCl3 (good solvent) and MeOH (poor solvent), afforded a benzannulated polymer.
Poly-3OTBS-tBu100
.
According to general procedure A, poly-3OTBS-tBu100 was obtained as an orange powder (94 mg) in 53% yield using poly-2OTBS (100 mg, Mn = 6400, PDI = 2.02), 1tBu (162 mg, 0.521 mmol) and AZADO (87 mg, 0.573 mmol): FT-IR (KBr): ν (cm−1) 3078, 3033, 2961, 2929, 2904, 2863, 1614, 1483, 1392, 1362, 1320, 1258, 1206, 1156, 1119, 1101, 1024, 1000, 952, 928, 892, 840, 813, 782, 734, 692, 653, 629. 1H NMR (500 MHz, CDCl3, 25 °C): δ (ppm) 8.61 (s, br, 4H), 8.0–6.0 (m, br, 14H), 1.6–1.0 (m, br, 66H). 13C NMR (125 MHz, CDCl3, 25 °C): δ (ppm) 148.9, 138.9, 131.3, 128.6, 124.5, 122.5, 34.9, 31.5, 26.2, 25.1, 18.5, 17.8, −4.00. 1H and 13C NMR spectra of poly-3OTBS-tBu100 are shown in Fig. S13 and S14 (ESI†), respectively.
Poly-3OTBS-tBu50
.
According to general procedure A, poly-3OTBS-tBu50 was obtained as an orange powder (98 mg) in 65% yield using poly-2OTBS (100 mg, Mn = 6400, PDI = 2.02), 1tBu (67 mg, 0.217 mmol) and AZADO (37 mg, 0.240 mmol): FT-IR (KBr): ν (cm−1) 3078, 3033, 2961, 2929, 2904, 2863, 2205 (vCC), 1906, 1792, 1684, 1615, 1506, 1485, 1473, 1443, 1395, 1363, 1335, 1258, 1204, 1117, 1102, 1004, 928, 890, 838, 815, 782, 735, 690, 620, 542. 1H NMR (500 MHz, CDCl3, 25 °C): δ (ppm) 8.70 (s, br, 4H), 7.70–6.58 (m, br, 22H), 1.56–0.42 (m, br, 92H). 13C NMR (125 MHz, CDCl3, 25 °C): δ (ppm) 150.0, 148.9, 138.3, 131.2, 127.8, 123.8, 122.8, 45.4, 37.5, 34.9, 31.4, 29.8, 27.5, 25.9, 25.1, 22.9, 21.4, 20.7, 19.9, 18.4, 17.7, 14.3, −4.2. 1H and 13C NMR spectra of poly-3OTBS-tBu50 are shown in Fig. S15 and S16 (ESI†), respectively.
Poly-3OTBS-H100
.
According to general procedure A, poly-3OTBS-H100 was obtained as an orange powder (95 mg) in 65% yield using poly-2OTBS (100 mg, Mn = 6400, PDI = 2.02), 1H (103 mg, 0.521 mmol) and AZADO (87 mg, 0.573 mmol): FT-IR (KBr): ν (cm−1) 3069, 3030, 2953, 2928, 2894, 2887, 2857, 1602, 1527, 1472, 1447, 1392, 1362, 1331, 1255, 1209, 1101, 1046, 1007, 981, 934, 884, 838, 804, 780, 758, 726, 700, 616, 658, 533. 1H NMR (500 MHz, CDCl3, 25 °C): δ (ppm) 8.80 (s, br, 4H), 7.69–6.65 (m, br, 4.6H), 1.71–0.41 (m, br, 8H). The solubility of the product was too low to record a reasonable 13C NMR spectrum. 1H NMR spectrum of poly-3OTBS-H100 is shown in Fig. S17 (ESI†).
Poly-3OHex-tBu100
.
According to general procedure B, poly-3OHex-tBu100 was obtained as an orange powder (150 mg) in 74% yield using poly-2OHex (100 mg, Mn = 10400, PDI = 4.50), 1tBu (186 mg, 0.600 mmol) and MnO2 (57 mg, 0.66 mmol): FT-IR (KBr): ν (cm−1) 3075, 3028, 2966, 2906, 2868, 1656, 1604, 1486, 1394, 1363, 1261, 1201, 1119, 1021, 1000, 894, 814, 735, 680, 612, 530. 1H NMR (500 MHz, CDCl3, 25 °C): δ (ppm) 8.67 (s, br, 4H), 7.71–6.55 (m, br, 14H), 3.60 (s, br, 4H), 1.59–0.24 (m, br, 58H). 13C NMR (125 MHz, CDCl3, 25 °C): δ (ppm) 147.6, 135.9, 127.5, 121.8, 66.7, 43.4, 35.5, 32.8, 29.4, 27.8, 25.4, 23.6, 20.7, 19.3, 18.6, 17.9, 12.1. 1H and 13C NMR spectra of poly-3OHex-tBu100 are shown in Fig. S18 and S19 (ESI†), respectively.
Poly-3OHex-H100
.
According to general procedure B, poly-3OHex-H100 was obtained as an orange powder (115 mg) in 76% yield using poly-2OHex (100 mg, Mn = 10400, PDI = 4.50), 1H (123 mg, 0.620 mmol) and MnO2 (56 mg, 0.64 mmol): FT-IR (KBr): ν (cm−1) 3061, 3021, 2954, 2926, 2868, 2857, 1921, 1657, 1597, 1490, 1469, 1446, 1434, 1413, 1352, 1262, 1200, 1086, 1045, 1020, 951, 930, 861, 800, 760, 726, 700, 651, 618, 530. 1H NMR (125 MHz, CDCl3, 25 °C): δ (ppm) 8.80 (s, br, 4H), 7.69–6.41 (m, br, 19H), 3.49 (s, br, 4H), 1.65–0.47 (m, br, 24H). 13C NMR (500 MHz, CDCl3, 25 °C): δ (ppm) 130.1, 127.3, 122.6, 68.9, 32.1, 31.7, 29.8, 29.5, 29.2, 25.9, 25.6, 22.7, 14.2. 1H and 13C NMR spectra of poly-3OHex-H100 are shown in Fig. S20 and S21 (ESI†), respectively.
Poly-3OH-tBu100
.
Under argon at 25 °C, a THF solution of tetrabutylammonium fluoride (1.0 M, 0.31 mL, 0.31 mmol) was added to an ODCB solution (2.5 mL) of poly-3OTBS-tBu100 (100 mg), which was synthesized from poly-2OTBS (Mn = 5900, PDI = 2.32), and the reaction mixture was stirred for 2 h at 25 °C, poured into water (10 mL) and extracted with CHCl3. The organic layer was washed with water, and partially evaporated under reduced pressure until the volume of the solution was reduced to ∼2.5 mL. The resulting concentrated solution was poured into vigorously stirred MeOH (50 mL). The resulting orange precipitate was collected by filtration, washed with MeOH (5 mL × 3 times) and dried under reduced pressure. Repeated reprecipitation of the resultant solid, using CHCl3 (good solvent) and MeOH (poor solvent), afforded poly-3OH-tBu100 (54.5 mg) as an orange powder in 77% yield: FT-IR (KBr): ν (cm−1) 3551, 3076, 3030, 2962, 2903, 2867, 1907, 1776, 1720, 1661, 1615, 1574, 1486, 1461, 1403, 1395, 1363, 1324, 1290, 1261, 1198, 1116, 1018, 989, 918, 891, 838, 813, 783, 749, 733, 651, 635, 543, 523. 1H NMR (500 MHz, CDCl3, 25 °C): δ (ppm) 8.63 (br, 4H), 7.71–6.44 (br, 14H), 1.55–0.02 (br, 36H). 13C NMR (125 MHz, CDCl3, 25 °C): δ (ppm) 149.7, 139.0, 131.3, 128.5, 125.6, 122.7, 35.0, 31.4, 29.9. 1H and 13C NMR spectra of poly-3OH-tBu100 are shown in Fig. S22 and S23 (ESI†), respectively.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas “π-Figuration: Control of Electron and Structural Dynamism for Innovative Functions” (JP26102008 for T. F.) and KAKENHI (17H03020 for Y. S.) from the Japan Society for the Promotion of Science (JSPS). This work was also supported in part by the Cooperative Research Program “CORE Lab” of “Network Joint Research Center for Materials and Devices: Dynamic Alliance for Open Innovation Bridging Human, Environment and Materials” from MEXT, Japan. Y. S. also acknowledges support from the Asahi Glass Foundation. The authors would like to thank Suzukakedai Materials Analysis Division, Technical Department, Tokyo Institute of Technology, for their support with the NMR measurements.
Notes and references
-
(a) J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869 CrossRef CAS PubMed;
(b) K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724 CrossRef CAS PubMed;
(c) M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196 CrossRef CAS PubMed;
(d) P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232 CrossRef CAS PubMed;
(e) H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875 CrossRef CAS PubMed;
(f) N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675 RSC;
(g) Z. Yang, R. Guo, R. Malpass-Evans, M. Carta, N. B. McKeown, M. D. Guiver, L. Wu and T. Xu, Angew. Chem., Int. Ed., 2016, 55, 11499 CrossRef CAS PubMed;
(h) F. Ishiwari, T. Sato, H. Yamazaki, J. N. Kondo, S. Miyanishi, T. Yamaguchi and T. Fukushima, J. Mater. Chem. A, 2016, 4, 17655 RSC.
-
(a) M. O’Keeffe and O. M. Yaghi, Chem. Rev., 2012, 112, 675 CrossRef PubMed;
(b) M. L. Foo, R. Matsuda and S. Kitagawa, Chem. Mater., 2014, 26, 310 CrossRef CAS;
(c) T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734 CrossRef CAS PubMed;
(d) B. Li, H.-M. Wen, Y. Cui, W. Zhou, G. Qian and B. Chen, Adv. Mater., 2016, 28, 8819 CrossRef CAS PubMed;
(e) K. K. Tanabe and S. M. Cohen, Chem. Soc. Rev., 2011, 40, 498 RSC;
(f) W. Lu,
et al.
, Chem. Soc. Rev., 2014, 43, 5561 RSC;
(g) V. Stavila, A. A. Talin and M. D. Allendorf, Chem. Soc. Rev., 2014, 43, 5994 RSC.
-
(a) S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548 RSC;
(b) X. Feng, X. Dinga and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010 RSC.
-
(a) A. I. Cooper, Adv. Mater., 2009, 21, 1291 CrossRef CAS;
(b) U. H. F. Bunz, K. Seehafer, F. L. Geyer, M. Bender, I. Braun, E. Smarsly and J. Freudenberg, Macromol. Rapid Commun., 2014, 35, 1466 CrossRef CAS PubMed;
(c) L. Tan and B. Tan, Chem. Soc. Rev., 2017, 46, 3322 RSC.
-
(a) T. Masuda, Y. Iguchi, B.-Z. Tang and T. Higashimura, Polymer, 1988, 29, 2041 CrossRef CAS;
(b) K. Nagai, T. Masuda, T. Nakagawa, B. D. Freeman and I. Pinnau, Prog. Polym. Sci., 2001, 26, 721 CrossRef CAS;
(c) J. Liu, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2009, 109, 5799 CrossRef CAS PubMed.
-
(a) P. M. Budd, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib and C. E. Tattershall, Chem. Commun., 2004, 230 RSC;
(b) N. B. McKeown, P. M. Budd, K. J. Msayib, B. S. Ghanem, H. J. Kingston, C. E. Tattershall, S. Makhseed, K. J. Reynolds and D. Fritsch, Chem. – Eur. J., 2005, 11, 2610 CrossRef CAS PubMed;
(c) N. Du, H. B. Park, G. P. Robertson, M. M. Dal-Cin, T. Visser, L. Scoles and M. D. Guiver, Nat. Mater., 2011, 10, 372 CrossRef CAS PubMed;
(d) M. Carta, R. Malpass-Evans, M. Croad, Y. Rogan, J. C. Jansen, P. Bernardo, F. Bazzarelli and N. B. McKeown, Science, 2013, 339, 303 CrossRef CAS PubMed;
(e) M. Carta, R. Malpass-Evans, M. Croad, Y. Rogan, M. Lee, I. Rose and N. B. McKeown, Polym. Chem., 2014, 5, 5267 RSC;
(f) M. Carta, M. Croad, J. C. Jansen, P. Bernardo, G. Clarizia and N. B. McKeown, Polym. Chem., 2014, 5, 5255 RSC;
(g) E. Madrid, Y. Rong, M. Carta, N. B. McKeown, R. Malpass-Evans, G. A. Attard, T. J. Clarke, S. H. Taylor, Y.-T. Long and F. Marken, Angew. Chem., Int. Ed., 2014, 53, 10751 CrossRef CAS PubMed;
(h) F. Xia, M. Pan, S. Mu, R. Malpass-Evans, M. Carta, N. B. McKeown, G. A. Attard, A. Brew, D. J. Morgan and F. Marken, Electrochim. Acta, 2014, 128, 3 CrossRef CAS;
(i) Y. Rong, R. Malpass-Evans, M. Carta, N. B. McKeown, G. A. Attard and F. Marken, Electroanalysis, 2014, 26, 904 CrossRef CAS;
(j) C. Li, A. L. Ward, S. E. Doris, T. A. Pascal, D. Prendergast and B. A. Helms, Nano Lett., 2015, 15, 5724 CrossRef CAS PubMed;
(k) X. Weng, J. E. Baez, M. Khiterer, M. Y. Hoe, Z. Bao and K. J. Shea, Angew. Chem., Int. Ed., 2015, 54, 11214 CrossRef CAS PubMed.
-
(a) C. K. Narula and H. Nöth, J. Organomet. Chem., 1985, 131, 281 Search PubMed;
(b) S. Biswas, I. M. Oppel and H. F. Bettinger, Inorg. Chem., 2010, 49, 4499 CrossRef CAS PubMed.
- Y. Shoji, N. Tanaka, S. Muranaka, N. Shigeno, H. Sugiyama, K. Takenouchi, F. Hajjaj and T. Fukushima, Nat. Commun., 2016, 7, 12704 CrossRef PubMed.
- U. H. F. Bunz, Chem. Rev., 2000, 100, 1605 CrossRef CAS PubMed.
-
(a) H. Li, D. R. Powell, R. K. Hayashi and R. West, Macromolecules, 1998, 31, 52 CrossRef CAS;
(b) S.-B. Lei, K. Deng, Y.-L. Yang, Q.-D. Zeng, C. Wang, Z. Ma, P. Wang, Y. Zhou, Q.-L. Fan and W. Huang, Macromolecules, 2007, 40, 4552 CrossRef CAS.
- Since the retention times of the zigzag polymers in the GPC analysis became much longer than those of parent PPEs, the polystyrene-based molecular-weight analysis was not applicable to the zigzag polymers. Instead, we used size-exclusion chromatography–multi-angle laser light scattering (SEC–MALS) for evaluating the molecular weight and PDI values of the zigzag polymers such as poly-3OTBS-tBu100 (Fig. S3, ESI†). The results are summarized in Table S1 (ESI†).
-
(a) K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, P. A. Pierotti and J. Rouquerol, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS;
(b) J. Rouquerol, D. Avnir, C. W. Fairbridge, D. H. Everett, J. M. Haynes, N. Pernicone, J. D. F. Ramsay, K. S. W. Sing and K. K. Unger, Pure Appl. Chem., 1994, 66, 1739 CrossRef CAS.
- N. B. McKeown and P. M. Budd, Macromolecules, 2010, 43, 5163 CrossRef CAS.
- M. Tashiro and T. Yamato, J. Org. Chem., 1979, 44, 3037 CrossRef CAS.
- C. Song and T. M. Swager, Macromolecules, 2005, 38, 4569 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary scheme, table, figures and analytical data. See DOI: 10.1039/c7qm00582b |
|
| This journal is © the Partner Organisations 2018 |

 *a,
Minsu
Hwang
a,
Haruka
Sugiyama
a,
Fumitaka
Ishiwari
*a,
Minsu
Hwang
a,
Haruka
Sugiyama
a,
Fumitaka
Ishiwari






