DOI:
10.1039/C7QM00564D
(Research Article)
Mater. Chem. Front., 2018,
2, 468-474
A circularly polarized luminescent organogel based on a Pt(II) complex possessing phenylisoxazoles†
Received
7th December 2017
, Accepted 15th January 2018
First published on 15th January 2018
Abstract
A Pt(II)phenylbipyridine complex 1, possessing phenylisoxazoles, long alkyl chains, and chiral alkyl chains, was synthesized. Complex 1 formed a stacked assembly in chloroform, self-assembled in a cooperative manner in methylcyclohexane (MCH), and gelled in higher alcohols and dodecane. In the nucleation regime, 1 assembled via a Pt–Pt interaction, which was demonstrated by the metal–metal-to-ligand charge transfer (MMLCT) emission band of the assembly. In the elongation regime, 1 displayed aggregation-induced emission enhancement (AIEE) character. The assembly of 1 formed in MCH was chiroptically non-active, suggesting that the assembly was non-helical. Complex 1 also exhibited strong AIEE and circularly polarized luminescence (CPL) with an anisotropic factor (glum) of 0.011 in 1-decanol gel, indicating that a chiral assembly was formed in the gel.
Introduction
Circularly polarized luminescence (CPL) is an emission that has different intensities of right- and left-circularly polarized light.1 CPL has attracted research interest due to its potential application in circularly polarized lights, security inks, and chemical sensors.2 Recently, many examples of CPL-active organic compounds and polymers have been reported.3 Although most of these CPL properties have been investigated in organic solutions, CPL in condensed phases, such as solids, films, and gels, is desired for the application of CPL-active compounds.4
CPL-active organic compounds have chirality in their structure, in which the chiral skeletons of binaphthyls5 and helicenes6 are often employed for constructing CPL-active organic compounds, as well as helical polymers.7 Recently, chiral supramolecular systems exhibiting CPL have been investigated due to their advantage in stimuli-responsivity.8 Our group has reported CPL-active supramolecular stacked polymers that possess luminescent dyes and phenylisoxazole moieties.9 A Pt(II)phenylbipyridine complex, possessing phenylisoxazoles, exhibits CPL with a high emission quantum yield (φem) and CPL anisotropic factor (glum).9b Although photo-functional organogels based on phenylisoxazoles have been investigated,10 CPL-active organogels based on phenylisoxazoles have not been developed due to the chiral 3,7-dimethyloctyl side-chain reduction in the intermolecular interaction, interfering with the formation of organogels.11 In this paper, we synthesize a novel Pt(II) phenylbipyridine complex 1 possessing a 3,5-bis(phenylisoxazolyl)phenylethynyl ligand. The introduction of four octadecyl side-chains was effective in constructing the organogels (Fig. 1).
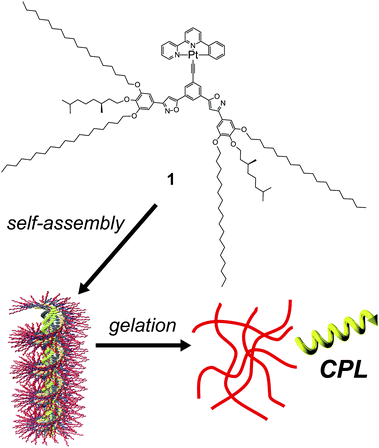 |
| | Fig. 1 Schematic representation of the self-assembly and CPL-active organogel of 1. | |
Results and discussion
Synthesis
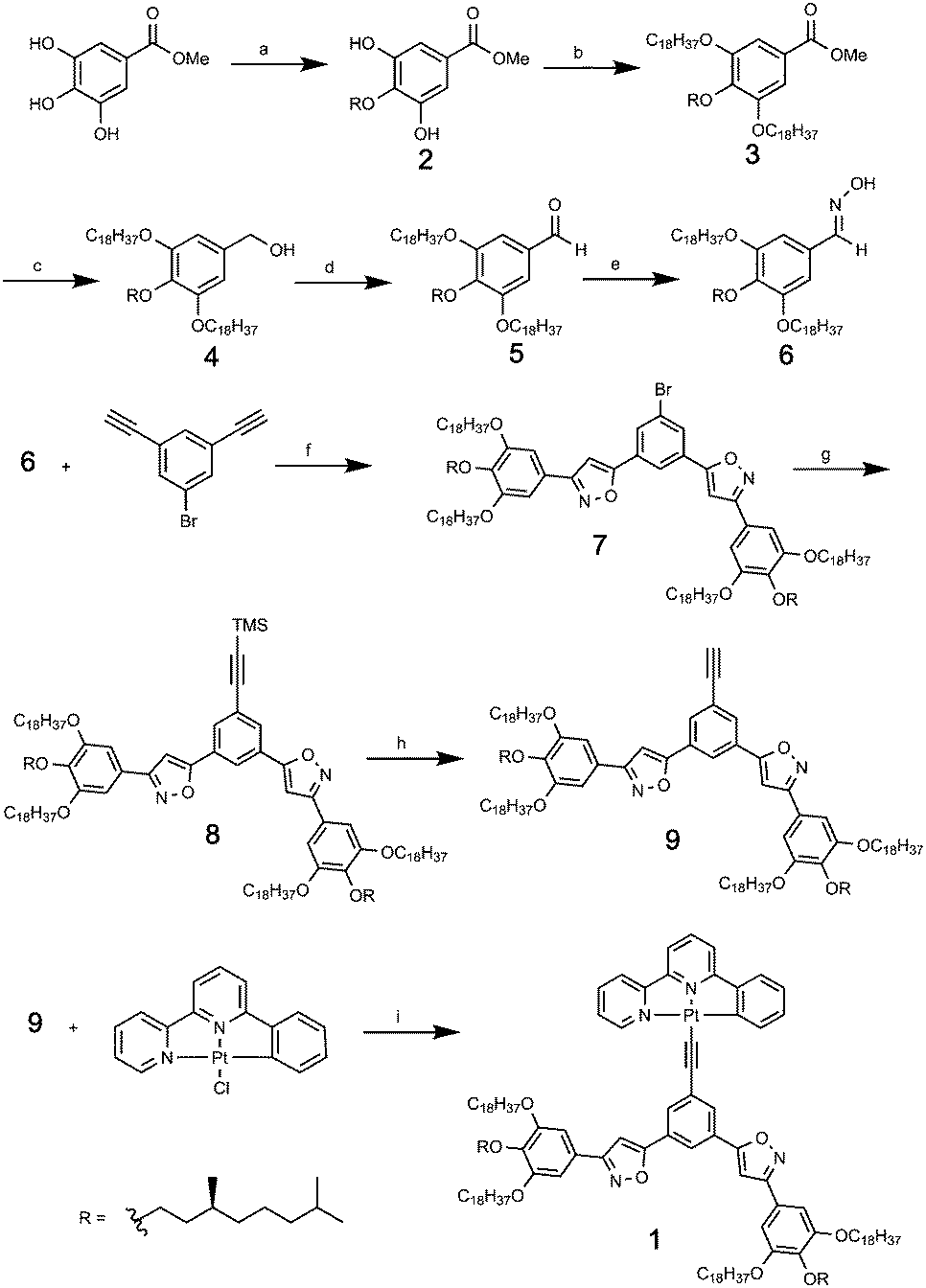
Compound 1 was synthesized as shown in Scheme 1. The central hydroxyl group of the three of methyl gallate was selectively alkylated by (S)-1-bromo-3,7-dimethyloctane in 50% yield to give 2. The remaining two hydroxyl groups were alkylated by using 1-bromooctadecane to afford 3 in 60% yield. After the conversion of methyl ester to oxime in 3 steps, a 1,3-dipolar addition reaction between the nitrile oxide prepared in situ from the oxime 6 and 3,5-diethynylbromobenzene12 gave the 1-bromo-3,5-bis(phenylisoxazolyl)benzene derivative 7 in 61% yield. The bromine of 7 was converted to an ethynyl group in two steps to provide ligand 9. Finally, (phenylbipyridyl)Pt(II) chloride was reacted with ligand 9 to give the desired compound 1 in 42% yield.
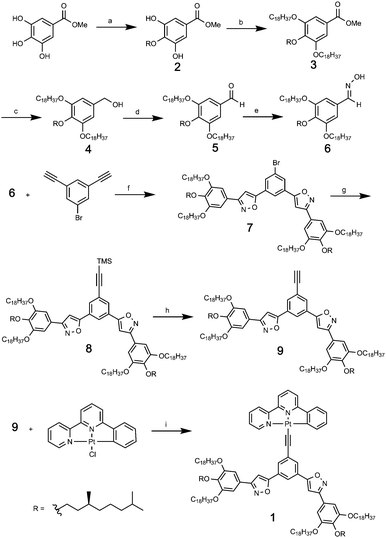 |
| | Scheme 1 Synthetic procedure for compound 1. (a) (S)-1-Bromo-3,8-dimethyloctane, K2CO3, dimethylformamide (DMF), 50%. (b) 1-Bromooctadecane, Cs2CO3, 2-butanone, 60%. (c) LiAlH4, tetrahydrofuran (THF), 97%. (d) MnO2, CH2Cl2, 98%. (e) NH2OH·HCl, NaOH, THF, H2O, 95%. (f) tBuOCl, NaI, 2,6-lutidine, 1,4-dioxane, 61%. (g) Trimethylsilyl acetylene, PdCl2(PPh3)2, CuI, iPr2NH, THF, 74%. (h) Tetrabutylammonium fluoride (TBAF), THF, 83%. (i) Triethylamine, CuI, CH2Cl, 42%. | |
Self-assembly in chloroform
The self-assembly behavior of 1 in solution was investigated using 1H NMR spectroscopy in chloroform-d (Fig. 2 and Fig. S1, ESI†). The aromatic protons of 1 were shifted upfield as the concentration of 1 increased from 0.71 to 14 mmol L−1. The upfield shift was explained by the formation of a stacked assembly; the aromatic protons of 1 were shielded by the ring current of the neighboring molecules. The plots of the chemical shifts of each proton vs. the concentration of 1 were used to make hyperbolic curves. The analysis, based on the isodesmic model,13 provides an association constant (Ki) of 36 ± 4 L mol−1 and association-induced shifts (Δδ) of −0.64, −0.28, −0.35, −0.39, −0.30, −0.14, −0.12, 0.01, and −0.02 ppm for protons Ha, Hb, Hc, Hd, He, Hf, Hg, Hh, and Hi, respectively. The protons of phenylbipyridine (Ha–He) shifted more than the protons of bis(phenylisoxazolyl)benzene (Hh and Hi), suggesting that 1 self-assembled as a pile around the Pt via a Pt–Pt interaction, in addition to π–π stacking and dipole–dipole interactions.
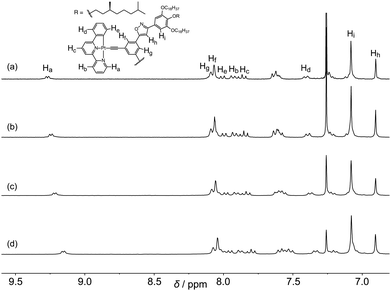 |
| | Fig. 2 Concentration-dependent 1H NMR spectra of 1 at 25 °C in chloroform-d. The concentrations of 1 are (a) 3.0, (b) 4.7, (c) 7.1, and (d) 14.2 mmol L−1. | |
Formation of the organogel
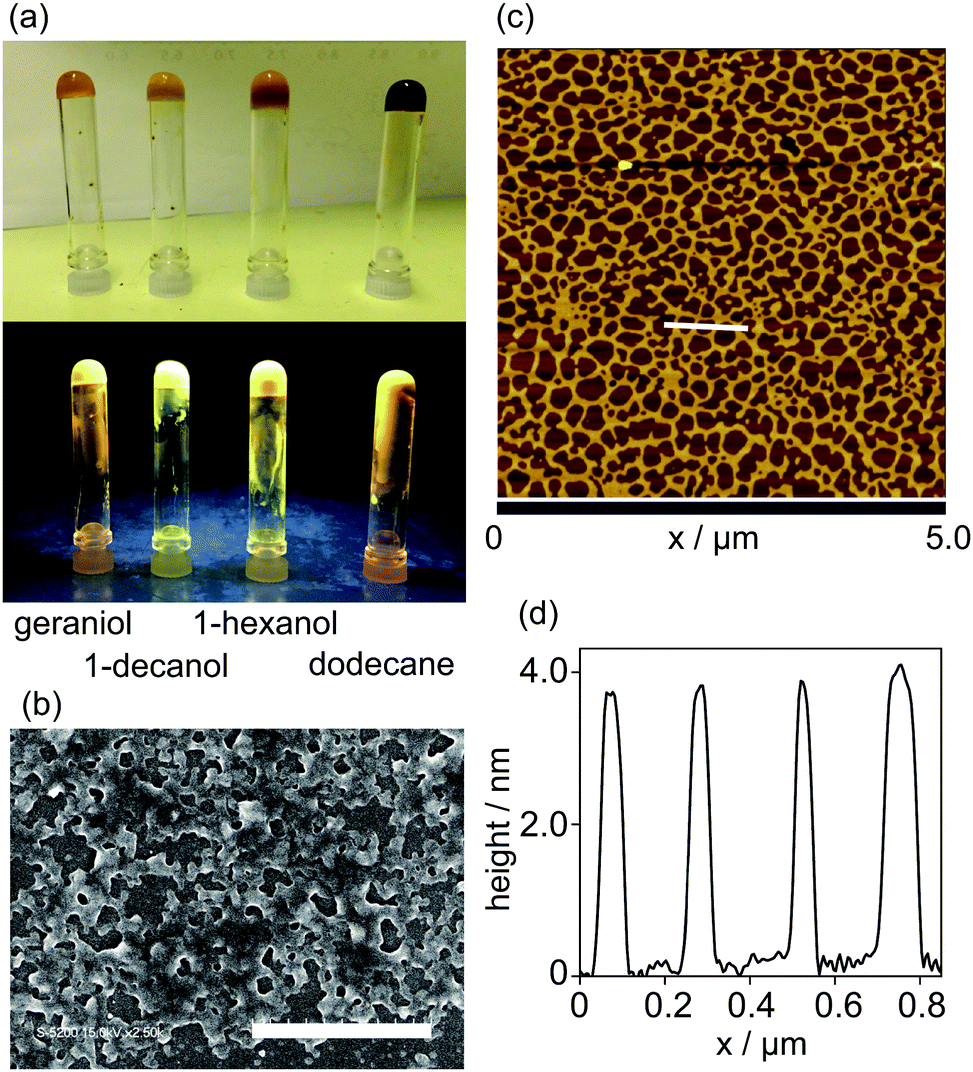
The gelation property of 1 was examined by the inverted test tube method, and the results summarized in Table 1. Complex 1 gelled in higher alcohols and dodecane, in which the alcohol gels were yellow and turbid, whereas the dodecane gel was dark yellow and turbid. The gels produced yellow luminescence under irradiation with UV light (Fig. 3a). The morphology of the 1-hexanol xerogel of 1 was observed using a field emission-scanning electron microscope (FE-SEM), and highly entangled networks with voids were observed (Fig. 3b). The solvent molecules are held in the voids, losing their fluidity in the gel. The fine morphologies of the assemblies were observed using an atomic force microscope (AFM). The sample was prepared by spin-coating a methylcyclohexane (MCH) solution of 1 ([1] = 1.5 × 10−5 mol L−1) onto a mica surface. Networked fibers with uniform height of ca. 4.0 nm, corresponding to the molecular size of 1, were observed (Fig. 3c, d and Fig. S2, ESI†). Each fiber had a width of more than 50 nm, suggesting that one-dimensional stacked assemblies form bundles on the surface, most likely due to the hydrophobic interaction of the alkyl side-chains.
Table 1 Gelation properties of 1a,b,c
| Solvent |
Solvent |
|
G = gel, P = precipitation, S = solution, and I = insoluble.
P, I, and S are at [1] = 20 g L−1.
The critical gelation concentration (g L−1) is shown in parentheses.
|
| Ethanol |
I |
Diethyl ether |
S |
| 1-Propanol |
P |
1,4-Dioxane |
P |
| 1-Butanol |
P |
Benzene |
S |
| 1-Hexanol |
G(14) |
Toluene |
S |
| 1-Decanol |
G(12) |
Butanone |
P |
| 2-Ethyl-1-hexanol |
G(15) |
Cyclohexanone |
S |
| Geraniol |
G(19) |
Acetophenone |
S |
| 2-Methoxyethanol |
I |
Methylcyclohexane |
I |
| Dodecane |
G(40) |
Chloroform |
S |
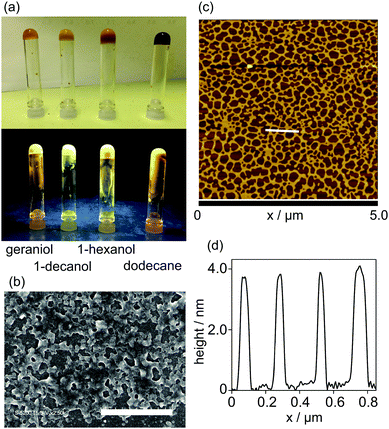 |
| | Fig. 3 (a) Photographs of organogels of 1 (top) under daylight and (bottom) under UV light. (b) The FE-SEM image of the xerogel of 1. Scale bar displays are 20 μm. (c) The AFM image of 1. (d) The height profile of the white line in (c). | |
Optical properties in solution
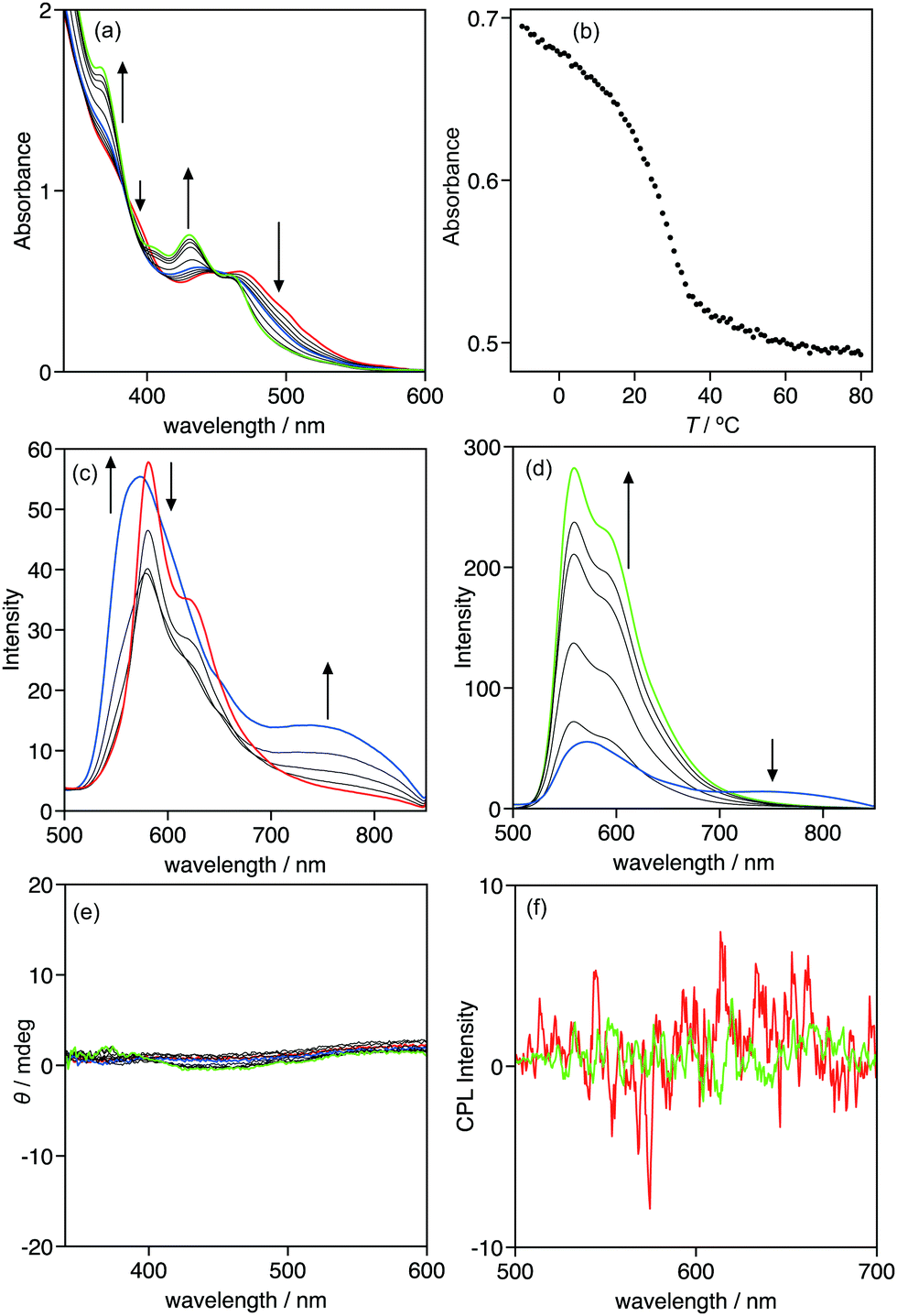
The optical properties of 1 were investigated using UV-vis absorption and emission spectroscopies. Complex 1 exhibited an absorption band at approximately 434 nm in chloroform (Fig. S3a, ESI†). Based on the association constant in chloroform-d and NMR estimation, most of complex 1 exists as a monomer in chloroform under the UV-vis conditions ([1] = 1.7 × 10−4 mol L−1). Thus, the absorption band observed at approximately 434 nm in chloroform is assignable to the metal–ligand-to-ligand charge transfer (MLLCT) absorption band of monomeric 1.9b,14 Changing the solvent to MCH, a less polar solvent than chloroform, resulted in the drastic change of the absorption spectra. Fig. 4a displays the absorption spectra of 1 in MCH at various temperatures ([1] = 1.5 × 10−4 mol L−1), in which they were temperature-dependent. At 80 °C, 1 exhibited a monomeric absorption band at approximately 444 nm. Decreasing the temperature triggered a two-step spectral change. From 80 to 50 °C, the absorption spectra changed, with isosbestic points at 382, 412, and 453 nm, and the MLLCT band hypsochromically shifted, suggesting that 1 self-assembled in an equilibrium system (Fig. S5, ESI†). Further cooling of the solution caused a spectral change without isosbestic points, indicating that the assembly process in this temperature range included more than two equilibrium systems. The plot of the absorbance at 430 nm vs. temperature clearly demonstrates a two-step assembly: the absorbance change was not sigmoidal and had a transition ca. 35 °C (Fig. 4b). This is a typical plot of a two-step assembly, known as a cooperative assembly, that includes an initial nucleation followed by an elongation regime.15 The established analysis of a cooperative assembly, based on the van der Schoot mathematical model,13,16 provides thermodynamic insights, including the elongation temperature (Te), the association constant in the elongation process at the elongation temperature (Ke(Te)), the enthalpic gain in the elongation process (he), the dimensionless equilibrium constant between the nucleation process and the elongation process (Ka), and the degree of polymerization at the elongation temperature (Nn(Te)). In the case of 1, the analysis of the elongation regime provided a Te of 310.4 K, he of −44.9 kJ mol−1, and Ke(Te) of 6.67 × 103 L mol−1 (Fig. S4, ESI†). The change in Gibbs free energy (ΔG) at Te was calculated from Ke(Te) to be −22.7 kJ mol−1 and the entropic change was calculated to be −71.5 J mol−1 K−1. These thermodynamic parameters indicate that the self-assembly in the elongation regime is an enthalpically driven and entropically opposed process. Pt–Pt, dipole–dipole, and π–π stacking interactions most likely provided the enthalpic gain. The analysis of the nucleation regime is difficult because the spectral change of the nucleation regime is completely different from that of the elongation regime, which prevents the estimation of Ka and Nn(Te).
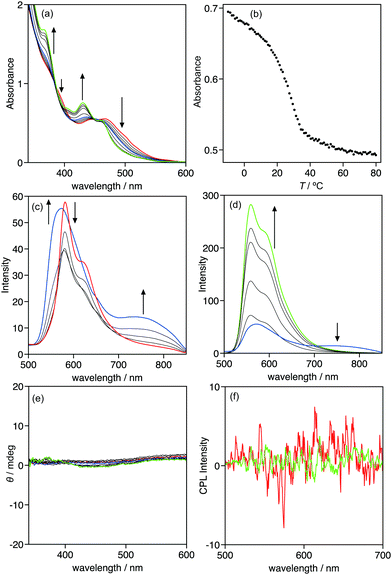 |
| | Fig. 4 (a) Temperature-dependent UV-vis absorption spectra of 1 in MCH. The spectra were recorded every 10 °C. The arrows indicate the change in the spectra as the temperature decreases from 80 to −10 °C. (b) Plot of the absorbance at 430 nm of 1 in MCH. (c and d) Temperature-dependent emission spectra of 1 in MCH. The spectra were recorded every 10 °C. The arrows indicate the change in the spectra as the temperature decreases from (c) 80 to 40 and (d) 40 to −10 °C. (e) Temperature-dependent CD spectra of 1 in MCH. The spectra were recorded every 10 °C from 80 to −10 °C. (f) Temperature-dependent CPL spectra of 1 in MCH at 80 and −10 °C. Red, blue, and green lines display the spectra at 80, 40, and −10 °C, respectively. [1] = 1.5 × 10−4 mol L−1; λex = 430 nm; l = 0.1 cm. | |
The emission spectra of 1 provided information about the structure of the assembly. In chloroform, 1 exhibits the monomeric emission assignable to a phosphorescence of the triplet metal-to-ligand charge transfer (3MLCT) band at 566 nm (Fig. S3b, ESI†).9b In MCH, the emission spectra of 1 were temperature-dependent, similar to the absorption spectra. At 80 °C, a 3MLCT band of a monomer was observed at 581 nm. The band decreased and a new band at 751 nm emerged as the temperature decreases from 80 to 50 °C, which is the nucleation regime. The new band is assignable to the metal–metal-to-ligand charge transfer (3MMLCT) emission band, suggesting that Pt–Pt interaction drives the assembly of 1 in the nucleation regime.9b,17 Further cooling of the solution triggered drastic change of the spectra: the 3MLCT band hypsochromically shifted to 559 nm, and emission was intensified ca. 5.2 times in the elongation regime. This intensification of the emission is known as aggregation-induced emission enhancement (AIEE).18 Restriction of the free rotation of a triple bond in the assembly may explain the AIEE of 1. It is noteworthy that the 3MMLCT emission band decreased in the elongation regime, despite the intensification of the 3MLCT band. The elongation of the π-conjugated molecule possessing phenylisoxazoles is driven by the multiple dipole–dipole interactions produced by the linear array of the dipole of the isoxazole.15c,d In the case of 1, the elongation may be driven by multiple dipole–dipole interactions, whereas the nucleation was driven by a Pt–Pt interaction.
Complex 1 has chiral alkyl side-chains, thus the assembly of 1 can exhibit chiroptical properties, including circular dichroism (CD) and CPL. However, the assembly of 1 in MCH did not display CD and CPL signals (Fig. 4e and f), suggesting that it is a non-helical assembly.
Optical properties of the gel
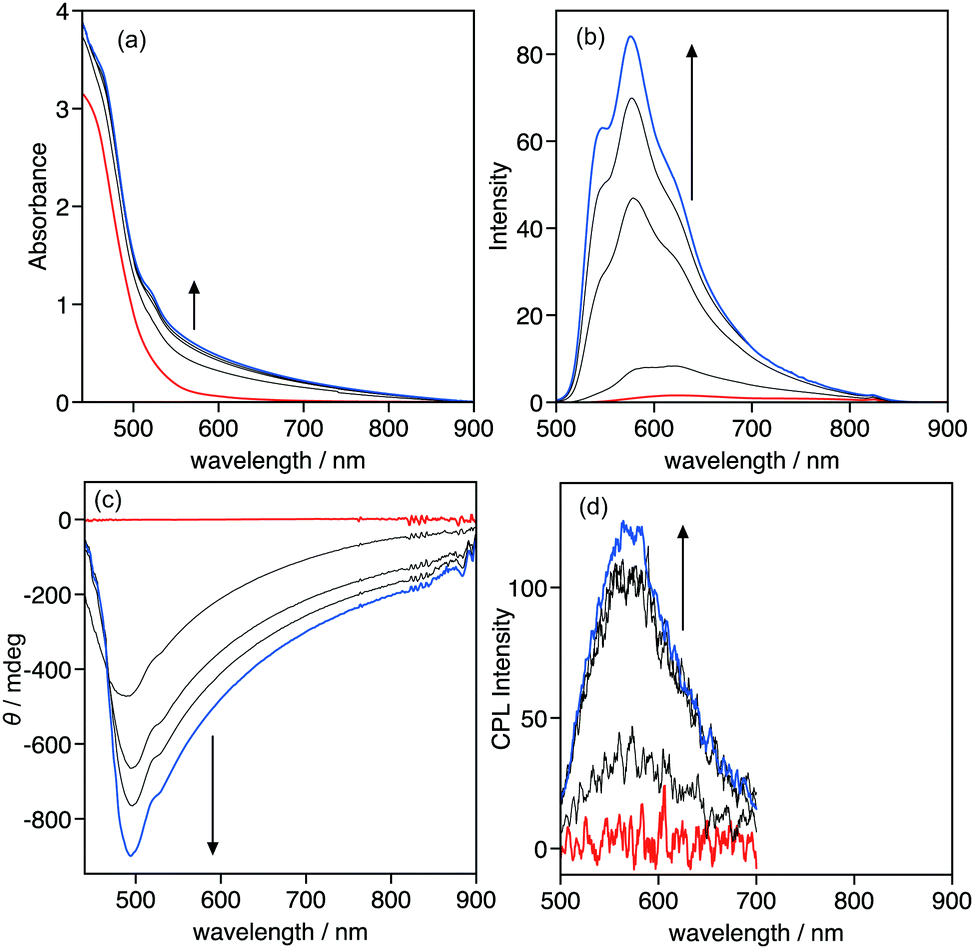
The optical and chiroptical properties of 1 in the gel phase are different from those in solution. Fig. 5 displays the UV-vis absorption, emission, CD, and CPL spectra of 1 in 1-decanol at various temperatures ([1] = 9.2 × 10−3 mol L−1). The gel–sol transition was observed at 41 °C under the experimental conditions, which was confirmed by heating the gel in the inverted test-tube. The UV-vis absorption and CD spectra of 1 under these conditions proved difficult to measure due to the high concentration, thus the absorption and CD spectra could be measured over 440 nm. At 50 °C, 1 was in a sol phase, displayed the foot of the MLLCT absorption band and no CD signals were observed. Cooling the sol from 50 to 10 °C caused the sol-to-gel transition that triggered the change in the absorption and CD spectra. The absorbance in the measured range gradually increased, and negative CD signals were observed below 40 °C, indicating that 1 formed a chiral helical assembly in the gel phase. Emission and CPL spectra also displayed a drastic change with the sol-to-gel transition. In the sol phase at 50 °C, the emission band of 1 was observed at 626 nm and no CPL signal was observed, which is the same as the CD result. The emission band was hypsochromically shifted to 576 nm and intensified during the sol-to-gel transition. The emission intensity increased ca. 50-fold from 50 to 0 °C. Positive CPL signals were observed upon the emission of 1 in the gel-phase with glum of 0.011. In the gel phase, the steric interaction between chiral side-chains became large compared to the solution phase, which may induce the helical chiral assembly of 1 in the gel phase to provide chiroptical properties, including CD and CPL.
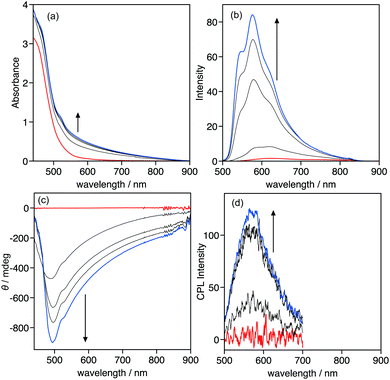 |
| | Fig. 5 Temperature-dependent (a) UV-vis absorption, (b) emission, (c) CD, and (d) CPL spectra of 1 in 1-decanol. The spectra were recorded every 10 °C. The arrows indicate the change in the spectra as the temperature decreases from 50 to 10 °C. Red and blue lines display the spectra at 50 and 10 °C, respectively. [1] = 9.2 × 10−3 mol L−1; λex = 430 nm; l = 0.1 cm. | |
Conclusions
In conclusion, a chiral low-molecular-weight organogelator 1 containing phosphorescent Pt(II)phenylbipyridine, phenylisoxazoles, chiral alkyl chains, and long alkyl chains was synthesized and the gelation and optical properties were investigated. Complex 1 formed a stacked assembly to produce a highly entangled network of fibers and gelled with higher alcohols and dodecane. Temperature-dependent UV-vis absorption and emission spectra in MCH solution reveal that 1 self-assembles in a cooperative manner. The emission of 1 is enhanced in the assembly compared to the monomer, which is a typical AIEE. Chiroptical properties are not observed in the solution, suggesting that the assembly is not helical in solution. In 1-decanol, CD and CPL signals are triggered by the sol-to-gel transition and helicity is induced by the gelation.
Experimental
General
All reagents and solvents were of the commercial reagent grade and were used without further purification unless otherwise noted. Dry CH2Cl2, DMF, and triethylamine were obtained by distillation over CaH2. 1H and 13C NMR spectra were recorded on a Varian mercury-300 spectrometer at 25 °C in chloroform-d and chemical shifts were reported as the delta scale in ppm relative to CHCl3 (δ = 7.260 for 1H and 77.3 for 13C). UV/vis absorption spectra were recorded on a JASCO V-560 spectrometer. Emission spectra were recorded on a JASCO FP-6500 spectrofluorometer. CD spectra were recorded on a JASCO J-1500 Circular Dichroism spectrometer. CPL spectra were recorded on a JASCO CPL-200 Circularly Polarized Luminescence spectrometer. ESI-Mass spectra were recorded on a Thermo Scientific LTQ Orbitrap XL hybrid FTMS. UV/vis absorption, emission, CD, and CPL spectra in solution were measured using a conventional quartz cell (light path 1 or 0.1 cm) with temperature control. UV/vis absorption, emission, CD, and CPL spectra in the gel phase were measured using a conventional quartz cell (light path 0.1 cm). Elemental analyses were performed using a CHN analyzer. Preparative separations were performed by silica gel gravity column chromatography (Silica Gel 60 N (spherical, neutral)). Recycling preparative GPC-HPLC separations were carried out on JAI LC-908s using preparative JAIGEL-2H, 2H, 1H columns in series.
Methyl 3,5-dihydroxy-4-((S)-3,7-dimethyloctyloxy)benzoate (2)
To a stirred solution of methyl gallate (5.00 g, 27.2 mmol) in 2-butanone (100 mL), (S)-1-bromo-3,7-dimethyloctane (3.84 g, 21.7 mmol) and Cs2CO3 (7.07 g, 21.7 mmol) were added. After being stirred at 80 °C for 21 h under an argon atmosphere, the resulting mixture was filtered and the filtrate was extracted with CH2Cl2. The organic layer was washed with water and brine, dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (CH2Cl2/hexane) to give 2 (3.21 g, 50%) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 7.22 (s, 2H), 5.44 (s, 2H), 4.17–4.10 (m, 2H), 3.88 (s, 3H), 1.89–1.78 (m, 1H), 1.69–1.44 (m, 2H), 1.37–1.08 (m, 7H), 0.94 (d, J = 7.2 Hz, 3H), and 0.87 (d, J = 7.2 Hz, 6H) ppm; 13C NMR (75 MHz, CDCl3): δ 166.5, 148.7, 137.6, 126.0, 109.7, 72.5, 52.2, 39.2, 37.3, 37.2, 29.8, 27.9, 24.6, 22.7, 22.6, and 19.6 ppm; HRMS (ESI+): calcd for C18H29O5m/z 325.2015 [M + H]+, found m/z 325.2021.
Methyl 4-((S)-3,7-dimethyloctyloxy)-3,5-bis(octadecyloxy)benzoate (3)
To a stirred solution of 2 (2.74 g, 8.44 mmol) in 2-butanone (60 mL), 1-bromooctadecane (6.14 g, 18.4 mmol) and Cs2CO3 (5.88 g, 18.0 mmol) were added. After being stirred at 80 °C for 21 h under an argon atmosphere, the resulting mixture was filtered and the filtrate was extracted with AcOEt. The organic layer was washed with water and brine, dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (AcOEt/hexane) to give 3 (4.21 g, 60%) as a white solid. M.p. 44–45 °C; 1H NMR (300 MHz, CDCl3): δ 7.25 (s, 2H), 4.09–3.98 (m, 6H), 3.89 (s, 3H), 1.87–1.74 (m, 6H), 1.57–1.09 (m, 68H), and 0.94–0.83 (m, 15H) ppm; 13C NMR (75 MHz, CDCl3): δ 166.6, 152.8, 142.4, 124.6, 107.9, 71.5, 69.0, 51.8, 39.4, 37.5, 37.4, 32.0, 29.8–29.3, 28.0, 26.2, 24.8, 22.7, 22.5, 19.5, and 14.1 ppm; HRMS (ESI+) calcd for C54H100O5Na m/z 851.7463 [M + Na]+, found m/z 851.7463; Anal. calcd for C54H100O5: C 78.20, H 12.15, found C 78.02, H 11.99%.
(4-((S)-3,7-Dimethyloctyloxy)-3,5-bis(octadecyloxy)phenyl)methanol (4)
To a stirred solution of LiAlH4 (474 mg, 12.5 mmol) in dry THF (30 mL) was added 3 (3.86 g, 4.66 mmol) in dry THF (20 mL) at 0 °C. After being stirred at room temperature for 1.5 h under an argon atmosphere, the reaction was quenched with saturated Na2SO4 aqueous solution. The resulting mixture was extracted with CHCl3. The organic layer was washed with brine, dried over anhydrous Na2SO4 and concentrated in vacuo to give 4 (3.61 g, 97%) as a white solid. M.p. 38–39 °C; 1H NMR (300 MHz, CDCl3): δ 6.56 (s, 2H), 4.59 (br, 2H), 4.02–3.90 (m, 6H), 1.87–1.74 (m, 6H), 1.57–1.09 (m, 68H), and 0.94–0.83 (m, 15H) ppm; 13C NMR (75 MHz, CDCl3): δ 152.9, 136.9, 136.6, 104.9, 71.6, 68.9, 64.8, 39.4, 37.6, 37.4, 32.0, 29.8–29.3, 28.0, 26.2, 26.2, 24.8, 22.7, 22.6, 19.5, and 14.1 ppm; HRMS (ESI+) calcd for C53H100O4Na m/z 823.7514 [M + Na]+, found m/z 823.7511; anal. calcd for C53H100O4: C 79.44, H 12.58, found C 79.21, H 12.46%.
4-((S)-3,7-Dimethyloctyloxy)-3,5-bis(octadecyloxy)benzaldehyde (5)
To a stirred solution of 4 (3.29 g, 4.10 mmol) in CH2Cl2 (100 mL) was added MnO2 (3.67 g, 42.3 mmol). After being stirred at room temperature for 24 h under an argon atmosphere, the reaction mixture was filtered, and the filtrate was concentrated in vacuo to give 5 (3.21 g, 98%) as a white solid. M.p. 41–42 °C; 1H NMR (300 MHz, CDCl3): δ 9.83 (s, 1H), 7.08 (s, 2H), 4.16–4.01 (m, 6H), 1.87–1.74 (m, 6H), 1.57–1.09 (m, 68H), and 0.94–0.83 (m, 15H) ppm; 13C NMR (75 MHz, CDCl3): δ 190.6, 153.5, 143.7, 131.5, 107.6, 71.6, 69.0, 39.4, 37.4, 37.3, 32.0, 29.8–29.3, 28.0, 26.1, 24.8, 22.7, 22.6, 19.5, and 14.1 ppm; HRMS (ESI+) calcd for C53H99O4m/z 799.7540 [M + H]+, found m/z 799.7542; anal. calcd for C53H98O4: C 79.64, H 12.36, found C 79.51, H 12.27%.
4-((S)-3,7-Dimethyloctyloxy)-3,5-bis(octadecyloxy)benzaldoxime (6)
To a stirred solution of 5 (3.21 g, 4.02 mmol) in THF (90 mL) was added a solution of NaOH (372 mg, 9.31 mmol) and hydroxylamine hydrochloride (635 mg, 9.13 mmol) in water (30 mL). After being stirred at 50 °C for 46 h under an argon atmosphere, the reaction mixture was extracted with CHCl3. The organic layer was washed with brine, dried over anhydrous Na2SO4 and concentrated in vacuo to give 6 (3.12 g, 95%) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 8.76 (s, 1H), 8.04 (s, 1H), 6.78 (s, 2H), 4.08–3.96 (m 6H), 1.87–1.74 (m, 6H), 1.57–1.09 (m, 68H), and 0.94–0.83 (m, 15H) ppm; 13C NMR (75 MHz, CDCl3): δ 153.5, 150.5, 140.1, 127.0, 105.6, 71.9, 69.3, 39.4, 37.5, 37.3, 31.9, 29.8–29.3, 28.0, 26.1, 24.7, 22.7, 22.6, 19.6 and 14.1 ppm; HRMS (ESI+) calcd for C53H100NO4m/z 814.7647 [M + H]+, found m/z 814.7656.
1-Bromo-3,5-bis(4-((S)-3,7-dimethyloctyloxy)-3,5-bis(octadecyloxy)phenylisoxazolyl)benzene (7)
To a stirred solution of 6 (3.12 g, 3.83 mmol), 1-bromo-3,5-diethynylbenzene (135 mg, 6.58 mmol), NaI (766 mg, 5.11 mmol), and 2,6-lutidine (5.0 mL, 43 mmol) in 1,4-dioxane (460 mL) was added tert-butyl hypochlorite (433 μL, 3.83 mmol). After being stirred at room temperature for 48 h under an argon atmosphere, the reaction was quenched with a portion of water. The resulting mixture was extracted with CH2Cl2, and the organic layer was washed with brine, dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (benzene/hexane) to give 7 (759 mg, 61%) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 8.23 (t, J = 1.6 Hz, 1H), 8.04 (d, J = 1.6 Hz, 2H), 7.06 (s, 4H), 6.91 (s, 2H), 4.11–4.02 (m, 12H), 1.87–1.74 (m, 12H), 1.57–1.09 (m, 136H), and 0.94–0.83 (m, 30H) ppm; 13C NMR (75 MHz, CDCl3): δ 167.6, 163.3, 153.6, 140.0, 129.9, 129.8, 123.7, 123.4, 118.7, 105.3, 99.2, 71.8, 69.3, 39.4, 37.5, 37.3, 31.9, 29.7–29.0, 28.0, 26.1, 24.8, 22.7, 22.6, 19.6, and 14.1 ppm; HRMS (APCI+) calcd for C116H200N2O8Br m/z 1828.4484 [M + H]+, found m/z 1828.4483.
1-((Trimethylsilyl)ethynyl)-3,5-bis(4-((S)-3,7-dimethyloctyloxy)-3,5-bis(octadecyloxy)phenylisoxazolyl)benzene (8)
To a stirred solution of 7 (759 mg, 0.415 mmol) in THF (30 mL), diisopropylamine (10 mL) and CuI (32.2 mg, 0.169 mmol) were added. The mixture was deoxygenated by bubbling nitrogen for 30 min, and then PdCl2(PPh3)2 (50.4 mg, 0.0718 mmol) and (trimethylsilyl)acetylene (2.0 mL, 14 mmol) were added to the reaction mixture. The reaction mixture was refluxed in the dark for 16 h under an argon atmosphere. The resulting mixture was poured into saturated aqueous ammonium chloride and extracted with CH2Cl2. The organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (benzene/hexane) to give 8 (569 mg, 74%) as a colorless oil. 1H NMR (300 MHz, CDCl3): δ 8.23 (t, J = 1.6 Hz, 1H), 7.98 (d, J = 1.6 Hz, 2H), 7.08 (s, 4H), 6.92 (s, 2H), 4.15–4.01 (m, 12H), 1.87–1.74 (m, 12H), 1.57–1.09 (m, 136H), 1.01–0.83 (m, 30H), and 0.33 (s, 9H) ppm; 13C NMR (75 MHz, CDCl3): δ 168.3, 163.2, 153.6, 134.0, 130.2, 128.5, 125.2, 123.5, 122.5, 105.3, 102.9, 98.8, 97.1, 71.8, 69.3, 39.4, 37.5, 37.4, 31.9, 29.8–29.3, 28.0, 26.1, 24.8, 22.7, 22.6, 19.6, 14.1, and −0.2 ppm; HRMS (ESI+) calcd for C121H209N2O8Si m/z 1846.5778 [M + H]+, found m/z 1846.5793.
1-Ethynyl-3,5-bis(4-((S)-3,7-dimethyloctyloxy)-3,5-bis(octadecyloxy)phenylisoxazolyl)benzene (9)
To a stirred solution of 8 (569 mg, 0.308 mmol) in THF (30 mL) was added tetrabutylammonium fluoride (221 mg, 0.699 mmol). After being stirred at room temperature for 45 min under an argon atmosphere, the reaction was quenched with a portion of water. The resulting mixture was extracted with Et2O. The organic layer was washed with saturated aq. NH4Cl and brine, dried over anhydrous Na2SO4, and concentrated in vacuo to give 9 as a colorless oil (459 mg, 83%). 1H NMR (300 MHz, CDCl3): δ 8.15 (t, J = 1.6 Hz, 1H), 7.89 (d, J = 1.6 Hz, 2H), 6.97 (s, 4H), 6.82 (s, 2H), 4.12–4.01 (m, 12H), 3.14 (s, 1H), 1.87–1.74 (m, 12H), 1.57–1.09 (m, 136H), and 0.94–0.83 (m, 30H) ppm; 13C NMR (75 MHz, CDCl3): δ 168.2, 163.2, 153.6, 140.0, 130.4, 128.6, 124.2, 123.5, 122.9, 105.3, 98.9, 81.7, 79.5, 71.8, 69.3, 39.4, 37.5, 37.4, 31.9, 29.7–29.2, 28.0, 26.1, 24.8, 22.7, 22.6, 19.6, and 14.1 ppm; HRMS (ESI+) calcd for C118H201N2O8m/z 1774.5383 [M + H]+, found m/z 1774.5390.
(6-Phenyl-2,2′-bipyridine)(3,5-bis(4-((S)-3,7-dimethyloctyloxy)-3,5-bis(octadecyloxy)phenylisoxazolyl)phenylethynyl)platinum (1)
To a stirred solution of 9 (450 mg, 0.254 mmol) and (6-phenyl-2,2′-bipyridine)platinum chloride (93.5 mg, 0.209 mmol) in CH2Cl2 (70 mL) and DMF (3.5 mL) was added triethylamine (3.5 mL). The reaction mixture was deoxygenated by bubbling nitrogen for 30 min, and then CuI (2.1 mg, 0.011 mmol) was added. After being stirred at room temperature for 17 h under an argon atmosphere in the dark, a portion of water was added and the resulting mixture was extracted with CH2Cl2. The organic layer was washed with brine, dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (CH2Cl2/hexane) to give 1 (215 mg, 42%) as an orange solid. M.p. 125–126 °C; 1H NMR (300 MHz, CDCl3): δ 9.25 (d, J = 6.4 Hz, 1H), 8.07 (t, J = 6.4 Hz, 1H), 8.07 (s, 2H), 8.06 (s, 1H), 8.00 (d, J = 7.5 Hz, 1H), 7.92 (d, J = 7.5 Hz, 1H), 7.85 (t, J = 7.5 Hz, 1H), 7.65–7.57 (m, 3H), 7.39 (d, J = 6.4 Hz, 1H), 7.24 (d, J = 7.5 Hz, 1H), 7.12–7.06 (m, 5H), 6.90 (s, 2H), 4.12–4.00 (m, 12H), 1.92–1.68 (m, 12H), 1.57–1.09 (m, 136H), and 0.97–0.83 (m, 30H) ppm; 13C NMR (75 MHz, CDCl3): δ 169.5, 168.4, 165.4, 163.1, 158.2, 154.4, 153.5, 151.6, 146.7, 141.9, 139.8, 138.9, 138.8, 132.8, 131.5, 130.3, 127.9, 127.6, 123.9, 123.8, 123.5, 122.7, 119.2, 118.5, 117.8, 105.3, 104.6, 98.4, 84.0, 80.5, 71.8, 69.2, 39.4, 37.5, 37.4, 31.9, 29.7–29.3, 28.0, 26.2, 24.8, 22.7, 22.7, 22.6, 19.6, and 14.1 ppm; HRMS (ESI+) calcd for C134H210N4O8NaPt m/z 2221.5689 [M + Na]+, found m/z 2221.5718; anal. calcd for C134H210N4O8Pt·H2O: C 72.56, H 9.63, N 2.53, found C 72.31, H 9.55, N 2.62%.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research (B, C), JSPS KAKENHI Grant Numbers JP15H03817, and JP15KT0145 from the Japan Society for the Promotion of Science (JSPS); by Grant-in-Aids for Scientific Research on Innovative Areas, JSPS KAKENHI Grant Numbers JP15H00946 (Stimuli-responsive Chemical Species), JP15H00752 (New Polymeric Materials Based on Element-Blocks), JP17H05375 (Coordination Asymmetry), and JP17H05159 (π-Figuration). Funding from the Iketani Science and Technology Foundation is gratefully acknowledged.
References
- J. P. Riehl and F. S. Richardson, Chem. Rev., 1986, 86, 1 CrossRef CAS.
-
(a) H. Goto and K. Akagi, Angew. Chem., Int. Ed., 2005, 44, 4322 CrossRef CAS PubMed;
(b) T. Uchida, K. Nozaki and M. Iwamura, Chem. – Asian J., 2016, 11, 2415 CrossRef CAS PubMed;
(c) M. Górecki, F. Zinna, T. Biver and L. Di Bari, J. Pharm. Biomed. Anal., 2017, 144, 6 CrossRef PubMed.
-
(a) H. Maeda and Y. Bando, Pure Appl. Chem., 2013, 85, 1967 CrossRef CAS;
(b) J. Kumar, T. Nakashima and T. Kawai, J. Phys. Chem. Lett., 2015, 6, 3445 CrossRef CAS PubMed;
(c) E. M. Sánchez-Carnerero, A. R. Agarrabeitia, F. Moreno, B. L. Maroto, G. Muller, M. J. Ortiz and S. de la Moya, Chem. – Eur. J., 2015, 21, 13488 CrossRef PubMed.
-
(a) S. H. Chen, D. Katsis, A. W. Schmid, J. C. Mastrangelo, T. Tsutsui and T. N. Blanton, Nature, 1999, 397, 506 CrossRef CAS;
(b) J. N. Wilson, W. Steffen, T. G. McKenzie, G. Lieser, M. Oda, D. Neher and U. H. F. Bunz, J. Am. Chem. Soc., 2002, 124, 6830 CrossRef CAS PubMed;
(c) A. Satrijo, S. C. J. Meskers and T. M. Swager, J. Am. Chem. Soc., 2006, 128, 9030 CrossRef CAS PubMed;
(d) Y. Zhao, N. A. A. Rahirn, Y. Xia, M. Fujiki, B. Song, Z. Zhang, W. Zhang and X. Zhu, Macromolecules, 2016, 49, 3214 CrossRef CAS;
(e) T. Goto, Y. Okazaki, M. Ueki, Y. Kuwahara, M. Takafuji, R. Oda and H. Ihara, Angew. Chem., Int. Ed., 2017, 56, 2989 CrossRef CAS PubMed.
-
(a) H. Tsumatori, T. Nakashima and T. Kawai, Org. Lett., 2010, 12, 2362 CrossRef CAS PubMed;
(b) J. Kumar, T. Nakashima, H. Tsumatori and T. Kawai, J. Phys. Chem. Lett., 2014, 5, 316 CrossRef CAS PubMed.
-
(a) K. E. S. Phillips, T. J. Katz, S. Jockusch, A. J. Lovinger and N. J. Turro, J. Am. Chem. Soc., 2001, 123, 11899 CrossRef CAS PubMed;
(b) J. E. Field, G. Muller, J. P. Riehl and D. Venkataraman, J. Am. Chem. Soc., 2003, 125, 11808 CrossRef CAS PubMed;
(c) T. Kaseyama, S. Furumi, X. Zhang, K. Tanaka and M. Takeuchi, Angew. Chem., Int. Ed., 2011, 50, 3684 CrossRef CAS PubMed;
(d) H. Oyama, K. Nakano, T. Harada, R. Kuroda, M. Naito, K. Nobusawa and K. Nozaki, Org. Lett., 2013, 15, 2104 CrossRef CAS PubMed;
(e) C. Shen, E. Anger, M. Srebro, N. Vanthuyne, K. K. Deol, T. D. Jefferson, G. Muller, J. A. G. Williams, L. Toupet, C. Roussel, J. Autschbach, R. Réau and J. Crassous, Chem. Sci., 2014, 5, 1915 RSC.
-
(a) S. Haraguchi, M. Numata, C. Li, Y. Nakano, M. Fujiki and S. Shinkai, Chem. Lett., 2009, 38, 254 CrossRef CAS;
(b) K. Watanabe, T. Sakamoto, M. Taguchi, M. Fujiki and T. Nakano, Chem. Commun., 2011, 47, 10996 RSC.
-
(a) F. C. Spano, S. C. J. Meskers, E. Hennebicq and D. Beljonne, J. Am. Chem. Soc., 2007, 129, 7044 CrossRef CAS PubMed;
(b) H. Maeda, Y. Bando, K. Shimomura, I. Yamada, M. Naito, K. Nobusawa, H. Tsumatori and T. Kawai, J. Am. Chem. Soc., 2011, 133, 9266 CrossRef CAS PubMed;
(c) A. Gopal, M. Hifsudheen, S. Furumi, M. Takeuchi and A. Ajayaghosh, Angew. Chem., Int. Ed., 2012, 51, 10505 CrossRef CAS PubMed;
(d) J. Liu, H. Su, L. Meng, Y. Zhao, C. Deng, J. C. Y. Ng, P. Lu, M. Faisal, J. W. Y. Lam, X. Huang, H. Wu, K. S. Wong and B. Z. Tang, Chem. Sci., 2012, 3, 2737 RSC;
(e) X.-P. Zhang, V. Y. Chang, J. Liu, X.-L. Yang, W. Huang, Y. Li, C.-H. Li, G. Muller and X.-Z. You, Inorg. Chem., 2015, 54, 143 CrossRef CAS PubMed;
(f) S. Tanaka, K. Sato, K. Ichida, T. Abe, T. Tsubomura, T. Suzuki and K. Shinozaki, Chem. – Asian J., 2016, 11, 265 CrossRef CAS PubMed.
-
(a) T. Ikeda, T. Masuda, T. Hirao, J. Yuasa, H. Tsumatori, T. Kawai and T. Haino, Chem. Commun., 2012, 48, 6025 RSC;
(b) T. Ikeda, M. Takayama, J. Kumar, T. Kawai and T. Haino, Dalton Trans., 2015, 44, 13156 RSC.
-
(a) T. Haino and H. Saito, Aust. J. Chem., 2010, 63, 640 CrossRef CAS;
(b) T. Haino, Y. Hirai, T. Ikeda and H. Saito, Org. Biomol. Chem., 2013, 11, 4164 RSC;
(c) H. Adachi, Y. Hirai, T. Ikeda, M. Maeda, R. Hori, S. Kutsumizu and T. Haino, Org. Lett., 2016, 18, 924 CrossRef CAS PubMed;
(d) T. Ikeda, T. Masuda, M. Takayama, H. Adachi and T. Haino, Org. Biomol. Chem., 2016, 14, 36 RSC;
(e) T. Ikeda and T. Haino, Polymer, 2017, 128, 243 CrossRef CAS;
(f) T. Ikeda, Y. Ueda, N. Komori, M. Abe and T. Haino, Supramol. Chem., 2017, 29, 471 CrossRef CAS.
-
(a) T. Haino, M. Tanaka and Y. Fukazawa, Chem. Commun., 2008, 468 RSC;
(b) M. Tanaka, T. Ikeda, J. Mack, N. Kobayashi and T. Haino, J. Org. Chem., 2011, 76, 5082 CrossRef CAS PubMed.
- S. Asmus, S. Beckendorf, M. Zurro, C. Mück-Lichtenfeld, R. Fröhlich and O. G. Mancheño, Chem. – Asian J., 2014, 9, 2178 CrossRef CAS PubMed.
-
Supramolecular Polymers, ed. A. Ciferri, CRC Press -Taylor & Francis, Boca Raton, FL, 2nd edn, 2005 Search PubMed.
- T. Moriuchi, Y. Sakamoto, S. Noguchi, T. Fujiwara, S. Akine, T. Nabeshima and T. Hirao, Dalton Trans., 2012, 41, 8524 RSC.
-
(a) P. Jonkheijm, P. van
der Schoot, A. P. H. J. Schenning and E. W. Meijer, Science, 2006, 313, 80 CrossRef CAS PubMed;
(b) T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687 CrossRef CAS PubMed;
(c) T. Ikeda, T. Iijima, R. Sekiya, O. Takahashi and T. Haino, J. Org. Chem., 2016, 81, 6832 CrossRef CAS PubMed;
(d) T. Ikeda, H. Adachi, H. Fueno, K. Tanaka and T. Haino, J. Org. Chem., 2017, 82, 10062 CrossRef CAS PubMed.
- M. M. J. Smulders, M. M. L. Nieuwenhuizen, T. F. A. de Greef, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Chem. – Eur. J., 2010, 16, 362 CrossRef CAS PubMed.
-
(a) M.-Y. Yuen, V. A. L. Roy, W. Lu, S. C. F. Kui, G. S. M. Tong, M.-H. So, S. S.-Y. Chui, M. Muccini, J. Q. Ning, S. J. Xu and C.-M. Che, Angew. Chem., Int. Ed., 2008, 47, 9895 CrossRef CAS PubMed;
(b) W. Lu, Y. Chen, V. A. L. Roy, S. S.-Y. Chui and C.-M. Che, Angew. Chem., Int. Ed., 2009, 48, 7621 CrossRef CAS PubMed.
-
(a) R. Hu, N. L. C. Leung and B. Z. Tang, Chem. Soc. Rev., 2014, 43, 4494 RSC;
(b) J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectral data, 1H and 13C NMR spectra. See DOI: 10.1039/c7qm00564d |
|
| This journal is © the Partner Organisations 2018 |

 ,
Kyohei
Hirano
and
Takeharu
Haino
,
Kyohei
Hirano
and
Takeharu
Haino