
A chemical sensor for CBr4 based on quasi-2D and 3D hybrid organic–inorganic perovskites immobilized on TiO2 films
 *a
and
Ioannis
Koutselas
*a
*a
and
Ioannis
Koutselas
*a
Abstract
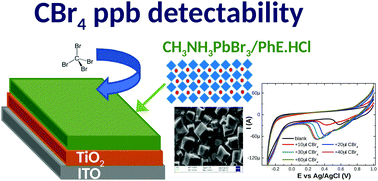
The effective immobilization of hybrid organic–inorganic semiconductors (HOIS) or their blends over standard mesoporous, transparent, semiconducting TiO2 films in order to study their electrochemical behavior as sensors through a cyclic voltammetry technique (CV) has been reported for the first time. The CV technique is used in order to acquire a comprehensive understanding of the electron transport dynamics within the TiO2 film, loaded with perovskites, while the structural and optical properties can be easily extracted from X-ray diffraction, optical absorption and photoluminescence measurements. The electrochemical behavior of the adsorbed perovskites was studied as a function of the potential applied to the semiconductive film, which induces transition from an insulating to a conductive state. The localized traps in the bandgap of the composite semiconductive film predominantly govern the electron transfer process. The HOIS employed are either 3D or mixtures of quasi-2D perovskites, for which there is strong evidence that the lead bromide and chloride based HOIS exhibit cathodic peaks at positive voltages. In addition, exploiting the capability of ions to migrate and interact with the HOIS lattice, a perovskite-based electrode was successfully used as an (electro-)chemical sensor for CBr4. All electrochemical processes concurring on the composite hybrid film vary with respect to the CBr4 concentration in the tested solution, leading to a CBr4 detectability range of the order of 20 ppb mol mol−1.
 Please wait while we load your content...
Please wait while we load your content...

