
Influence of alkyl chain length in S,N-heteropentacenes on the performance of organic solar cells†‡
Tanja D.
Leitner
a,
Astrid
Vogt
a,
Duško
Popović
a,
Elena
Mena-Osteritz
a,
Karsten
Walzer
b,
Martin
Pfeiffer
b and
Peter
Bäuerle
 *a
*a
aInstitute of Organic Chemistry II and Advanced Materials, University of Ulm, Albert-Einstein-Allee 11, 89081, Ulm, Germany. E-mail: peter.baeuerle@uni-ulm.de
bHeliatek GmbH, Treidlerstrasse 3, 01139, Dresden, Germany
First published on 26th February 2018
Abstract
A novel family of alkyl-substituted S,N-heteropentacenes SN5 4–6 with a systematic variation of the alkyl side chains from methyl to hexyl is presented. The parent heteroacene series 4 and the acceptor end-capped series 5 and 6 were synthesized and characterized with respect to their structure–property relationships. The dicyanovinyl (DCV) end-capped derivatives 6a–f were implemented as donors in vacuum-processed bulk-heterojunction solar cells with fullerene C60 as acceptor, their photovoltaic behaviour studied, and structure–device relationships discussed. The devices exhibited power conversion efficiencies (PCE) in the range of 4–6% and interestingly a correlation between the length of the alkyl side chains and the photovoltaic performance of the resulting devices was discovered: an odd–even effect with higher values for the even-numbered alkyl chains. Atomic force microscopy (AFM) studies on the active layer surfaces gave hints for the explanation of this effect.
1. Introduction
During the last years, research in the field of organic solar cells (OSC) made major steps forward as a result of combined efforts in the development of new organic semiconductors and the optimization of device fabrication.1 This progress recently culminated in the reported power conversion efficiency (PCE) of 13.1% in polymer-based fullerene-free OSCs.2 On the other hand, the utilization of oligomers comprising defined monodisperse structures and high reproducibility concerning their synthesis and device fabrication1a,b have manifested their important role in solution-processed bulk-heterojunction solar cells (BHJSC) from which devices with PCEs of ∼13% could be realized by using inverted device architectures.3 Another advantage over polymeric materials is the possibility of vacuum-deposition if a certain thermal stability of the utilized organic components is given.4 The potential of vacuum-processing was demonstrated with multi-junction cells reaching PCEs of up to 13.2%.5 Furthermore, vacuum-deposition gives the opportunity to avoid the use of toxic chlorinated solvents, which are mandatory for highly efficient solution-processed devices, and allows the construction of complex multilayer device structures with highly controlled and well-defined layers of high purity.1a,6S,N-Heteroacenes became a well-established class of compounds among oligomeric donor materials, e.g. for BHJSCs, as a result of the synergy between the increased planarity with strong bathochromically shifted absorptions and higher stability compared to non-fused oligothiophenes and thienoacenes, respectively.7 Although the bridging nitrogen atoms give the opportunity of attaching various side chains, certain aspects have to be taken into account: (i) the length of linear or branched alkyl side chains, which primarily serve to counteract strong π–π interactions, influences the solubility of oligomers in common organic solvents which is mandatory to a certain extent concerning controlled synthesis and purification, and (ii) has a strong impact on the morphology and therefore the efficiency of the resulting devices. This complex interplay has been highlighted in recent studies concerning the impact of side chains for various π-systems.8
With respect to organic solar cells, Gadisa et al. reported PCEs of 3.2%, 4.3%, and 4.6% for active layers consisting of poly(3-alkylthiophenes) as donors comprising butyl, pentyl, or hexyl side chains and [6,6]-phenyl-C61-butyric acid methyl ester (PC61BM) as acceptor.9 Although the polymeric backbone remained the same, the efficiency of the resulting devices could be correlated to differences in fill factor (FF), which is indicative for changes in morphology. You and co-workers, who prepared six differently substituted low band gap polymers, proposed that short and branched side chains would lead to a favourable balance between short-circuit current density (JSC) and open circuit voltage (VOC) to reach the optimum efficiency.10 This finding was also evidenced by the group of Hill with the use of differently nitrogen-substituted perylene diimides as acceptors in oligomer-based OSCs, in which the ethylpropyl-substituted derivative of this series gave the highest PCE.11 Our group has recently shown that even the branching point along the alkyl side chain influences the performance of the resulting device.12 Although the investigated molecules share the same conjugated backbone and therefore the same optoelectronic properties, the spatial distance of the branching point from the π-system led to increased JSC values and therefore the PCE increased from 7.7% to 8.2%.
Terminally acceptor-functionalized S,N-heteroacenes were implemented in organic electronic devices and corresponding S,N-heterohexacenes (SN6) showed good charge transport properties in organic field effect transistors and excellent performance in BHJSCs reaching a PCE of 7.1%.7b,13 In this series, systematic structural variation of the alkyl side chains revealed that optical and redox properties, which depend on the length of the conjugated backbone, as expected remained almost unaffected, whereas a noticeable influence on thermal and photovoltaic properties was found. Despite the insulating nature of alkyl side chains, an odd–even effect of the photovoltaic parameters with respect to the alkyl chain length was detected.
As well the smaller homologues, S,N-heteropentacenes (SN5), turned out to be excellent donors and p-type semiconductors in vacuum-processed and in solution-processed BHJSCs reaching PCEs of 6.5%7a and 4.9%,14 respectively. Furthermore, SN5-derivatives were structurally fine-tuned and implemented as hole transport material in perovskite solar cells leading to efficiencies close to 17%.15
In this contribution, we report synthesis, characterization, and photovoltaic device performance of a series of dicyanovinyl (DCV) end-capped S,N-heteropentacenes, in which the length of the alkyl side chains attached at the nitrogen atoms was systematically varied from methyl to hexyl. The presented acceptor-flanked oligomers 6a–f (Scheme 1) were used as p-type and C60 as n-type organic semiconductor in vacuum-processed BHJSCs giving devices with PCEs in the range of 4–6%. Interesting structure–device relationships were discovered by means of a noticeable odd–even effect with higher values for the even-numbered alkyl chains which is in contrast to the corresponding S,N-heterohexacenes. In order to understand the impact of the length effect of the alkyl chains and the odd–even effect, investigations of the surface of the photoactive layer by means of and atomic force microscopy (AFM) were undertaken.
 | ||
| Scheme 1 Synthesis of the SN5-series 4a–f, 5a–f, and DCV-substituted 6a–f. | ||
2. Results and discussion
2.1 Synthesis
Acceptor-substituted S,N-heteropentacenes 6a–6f were synthesized by a Knoevenagel condensation reaction of dialdehydes 5a–5f with malononitrile in excellent yields between 77% and 99% (Scheme 1).7a The crucial building block 3,3′,3′′,4′-tetrabromo-2,2′:5′,2′′-terthiophene 316 was synthesized in 76% yield by Pd-catalyzed Negishi-type cross-coupling reaction of tetrabromothiophene 1 and (3-bromothien-2-yl)zinc(II) chloride 2. The next step was a fourfold Pd-catalyzed Buchwald–Hartwig amination of terthiophene 3 with corresponding primary amines. S,N-Heteropentacenes 4a–4f were obtained in good yields of 52–63% after column chromatography.16 The obtained fused heteroacenes 4a–4f were converted into corresponding dialdehydes 5a–5f by Vilsmeier–Haack formylation in 55–86% yield after column chromatography.Due to the different alkyl chain lengths and therefore different solubility of the molecules among the series, the purification methods had to be adapted in every reaction step leading to different yields within each series. After typical chromatography, all targeted DCV-substituted SN5-derivatives 6a–f were further treated by fractionated sublimation to remove minor impurities, which could hamper device performances.
2.2 Thermal properties
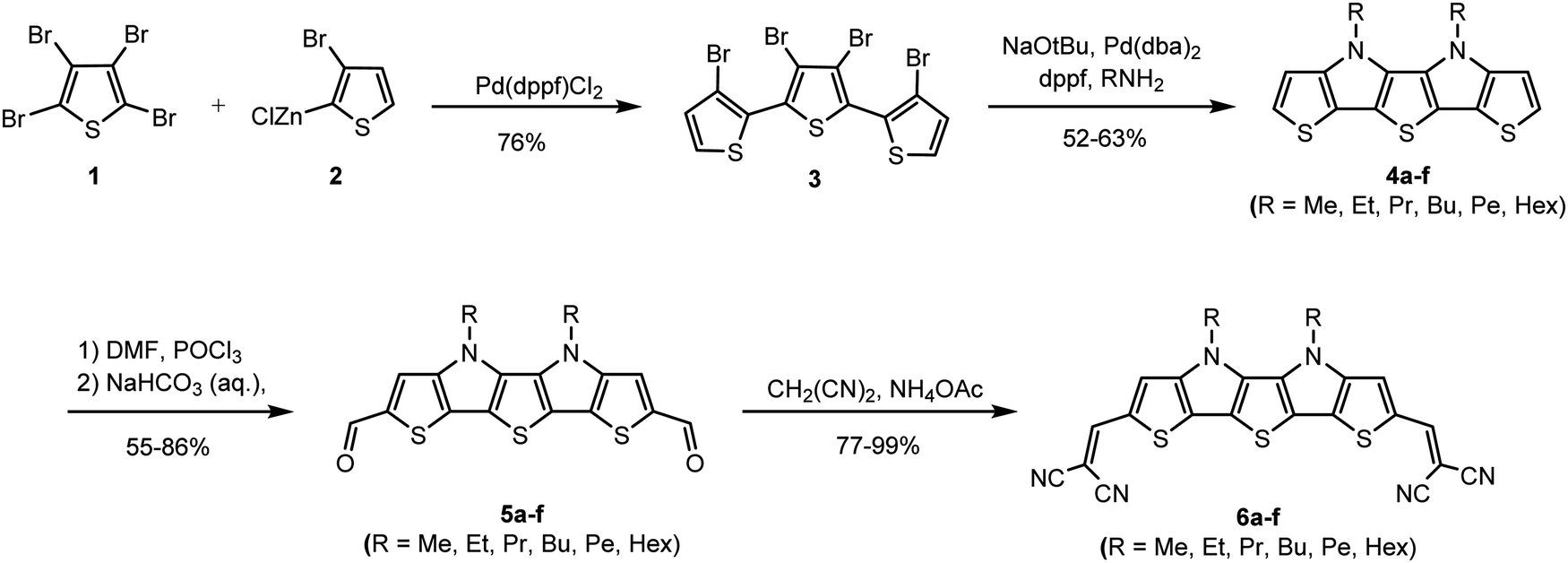
A high thermal stability of donor materials is very important for the construction of vacuum-deposited organic solar cells. Therefore, we investigated melting points or decomposition temperatures of the three series of SN5-derivatives 4–6 by differential scanning calorimetry (DSC) (ESI,‡ Fig. S1 and Table S1). As expected, within a series the melting points continuously decrease with increasing length of the alkyl side chains due to increased side chain disorder.17 Among the series, the DCV-substituted derivatives 6 exhibited higher melting points than the dialdehydes 5 and the parent SN5 4 due to the highest intermolecular interactions (Fig. 1).18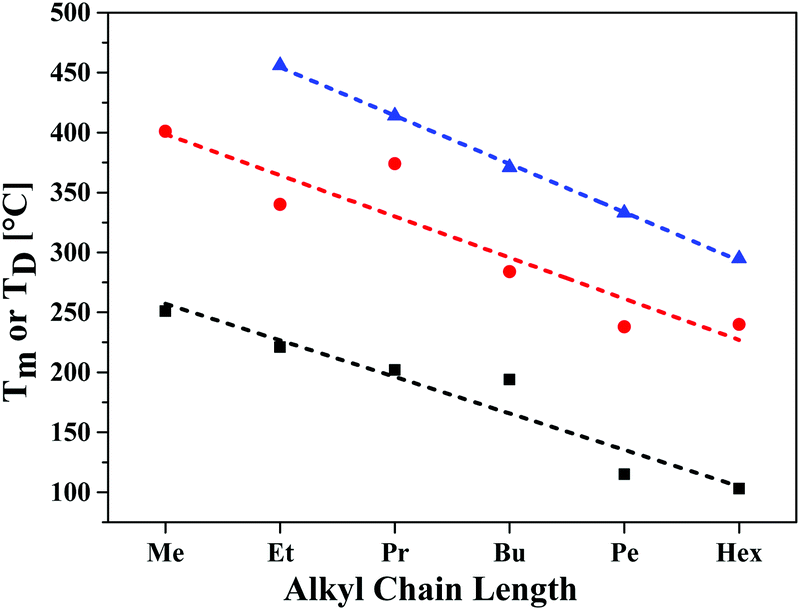 | ||
| Fig. 1 Graphic representation of melting points (Tm) or decomposition temperatures (TD) of SN5-derivatives 4a–f (black line), dialdehydes 5a–f (red line), and DCV-derivatives 6b–f (blue line). For methylated DCV-SN5 6a neither a melting point nor a decomposition temperature was measurable. | ||
In order to obtain very pure organic semiconductor materials for solar cell fabrication, we typically purify them by fractionated sublimation. As an example, the result for the propyl-substituted DCV-SN5 6c is shown in Fig. 2. The sublimation was performed at 340 °C source temperature and the pure product was isolated from fraction 1 showing exclusively the molecular ion peak at 510 amu in a high-resolution MALDI mass spectrum (HRMS, Fig. 3a). Minor impurities at higher masses, which were present before sublimation, were successfully removed. In fractions 2–5, however, increasing amounts of impurities formed during the sublimation process showing lower masses than product 6c were identified contaminating these product fractions. The formed impurities were isolated on a small scale and characterized by 1H- and 13C-NMR spectroscopy and by HRMS (Fig. 3b). The main mass peak at 448 amu we assign to the structure of a corresponding SN5-derivative comprising a methyl group instead of a DCV-group at one terminus. The minor mass peak at 434 amu correlates to a similar derivative having a proton instead of the methyl group at this position. The formation of such by-products in higher fractions of the sublimation was as well identified for the other DCV-SN5 derivatives. We could not yet elucidate the mechanism of the formation of these by-products, but it shows that the target molecules are not fully stable under the conditions present in the sublimation apparatus.
 | ||
| Fig. 2 Fractionated sublimation of propyl-substituted DCV-SN5 derivative 6c using a source temperature of 340 °C. The setup allows separate harvesting of different fractions, which re-crystallize from left to right in harvest rings with decreasing temperature, labelled as 1 to 5. | ||
 | ||
| Fig. 3 High-resolution MALDI mass spectra of (a) pure DCV-S,N-heteropentacene 6c after sublimation and (b) of by-products isolated by column chromatography after fractionated sublimation at 340 °C. Assigned structures are shown on the right. | ||
2.3 Optical and electrochemical properties
The influence of the different alkyl chain lengths on optical and electrochemical properties of the synthesized unsubstituted (4a–f), dialdehyde (5a–f), and DCV-substituted SN5-derivatives (6a–f) is as expected marginal (see ESI,‡ Table S1). Therefore, we exemplarily discuss the results of the pentyl-substituted derivatives 4e, 5e, and 6e. In the UV-vis absorption spectra taken in dichloromethane solution, we find a substantial red-shift of the main absorption band from 356 nm for 4e to 456 nm for 5e and to 582 nm for 6e according to an increased charge-transfer (CT) character of the transition. Concomitantly, the optical gap is gradually reduced from 3.35 eV for 4e to 2.00 eV for 6e and the molar extinction coefficient is (nearly) doubled derivative-to-derivative reaching a very high value of 152.700 M−1 cm−1 for the DCV-derivative 6e bearing the strongest acceptor group (Fig. 4, left). The increase in extinction coefficient is due to the increased transition probability of the CT-band due to increased acceptor strength. In Fig. 4 (right) the absorption and emission spectrum of DCV-derivative 6e in solution are shown in comparison to the absorption in thin film, which was prepared by evaporation. The absorption maximum of the main band (λmax = 588 nm) is only slightly red-shifted compared to the solution spectrum (λmax = 582 nm), but the band is significantly broadened leading to panchromatic absorption and reduced optical gap of 1.72 eV (Table 1). | ||
| Fig. 4 Left: UV-vis spectra of pentyl derivatives of parent 4e (black line), dialdehyde 5e (red line), and DCV-substituted 6e (blue line) measured in dichloromethane at room temperature. Right: UV-vis (black line) and fluorescence spectrum (blue line) of DCV-substituted 6e measured in dichloromethane at room temperature and UV-vis spectrum of a thin film of 6e evaporated on ITO (dashed line). | ||
The strong absorption combined with the lowered optical gap renders DCV-derivative 6e to a very promising candidate for light harvesting applications (vide infra). The emission maximum in solution at 619 nm leads to a relatively small Stokes shift around 1000 cm−1 indicating a small geometric change upon excitation due to the planar and stiff structure of the conjugated π-system.
The redox properties of the three series of SN5-derivatives 4–6 were determined by cyclic voltammetry in a dichloromethane or 1,2-dichloroethane/tetrabutylammonium hexafluorophosphate (0.1 M) electrolyte and the potentials were referenced against the redox couple ferrocene/ferricenium (Fc/Fc+), which was set to −5.1 eV vs. vacuum,19 in order to calculate the energetic levels of the frontier orbitals (HOMO, LUMO), which are important for solar cell application (ESI,‡ Table S1). The redox properties were as well only marginally influenced by the alkyl side chain length, therefore, pentyl-substituted SN5-derivatives 4e, 5e, and 6e were again taken as representatives for all studied derivatives with different chain lengths (Fig. 5, Table 2 and Table S1, ESI‡). Whereas the parent system 4e shows one irreversible oxidation peak at 0.14 V, acceptor-substituted 5e and 6e were reversibly oxidized at higher potentials (Eox1 = 0.55 V and 0.62 V) indicating the formation of stable radical cations. Quasi-reversible further oxidation to dications occurs at potentials higher than 1 V. According to the electron-withdrawing effect of the acceptor groups, the latter compounds were irreversibly reduced at −1.98 V and −1.53 V, respectively. The resulting electrochemical gaps follow the trend of the optical measurements. The energy levels of the frontier orbitals, which become important for the use of the materials in organic solar cells, were calculated from the onset value of the oxidation and reduction waves. As expected, in the series of pentyl-substituted SN5-derivatives 4e, 5e, and 6e HOMO and LUMO are gradually destabilized with increasing acceptor strength. For DCV-derivative 6e the HOMO energy was calculated to −5.65 eV, which is low enough to assure high open circuit voltages (VOC) in bulk-heterojunction solar cells, and the LUMO energy to −3.80 eV, which is sufficiently high to provide efficient electron transfer to the LUMO of the fullerene acceptor typically used in solar cells.7a,20
 | ||
| Fig. 5 Cyclic voltammograms of pentyl-substituted SN5-derivatives 4e (black line), 5e (red line), and 6e (blue line) measured in dichloromethane at room temperature, supporting electrolyte TBAPF6 (0.1 M), scan rate 100 mV s−1, potentials versus Fc/Fc+. | ||
2.4 Photovoltaic performance
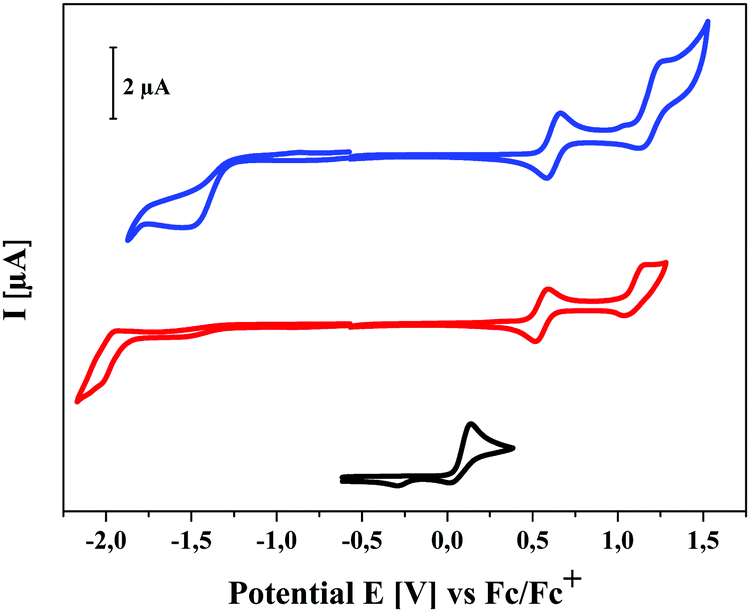
DCV-substituted S,N-heteropentacenes are well suited donor materials in vacuum-processed bulk-heterojunction solar cells. In a previous publication, we have disclosed that under optimized conditions with fullerene C60 as acceptor power conversion efficiency (PCE) of up to 6.5% can be reached with this class of heteroacenes. However, a decline of the performance was noted when the substituents at the SN5-nitrogens were varied from propyl (5.6%) to hexyl (3.7%) and to tolyl (0.7%) indicating that the photovoltaic parameters are dependent on the type and length of the alkyl side chains by influencing the morphology of the photoactive donor/acceptor layer.7a In order to better understand this influence of alkyl side chains in DCV-SN5 6a–f, bulk-heterojunction solar cells with m–i–p-type (metal-intrinsic-p-doped) device architecture were prepared by vacuum deposition.6 The DCV-SN5 derivatives 6a–f were used as donor (D) and C60 as acceptor (A) of the bulk-heterojunction. For the preparation of efficient organic solar cells, different parameters such as layer thickness, D–A ratio, or substrate temperature were varied. The order of the layer sequence in the devices was ITO/C60 (15 nm)/DCV-SN5 6a–f:C60 (20–30 nm)/BPAPF (9,9-bis[4-(N,N-bis-biphenyl-4-yl-amino)phenyl]-9H-fluorene, 10 nm)/BPAPF:NDP9 (45 nm, ca. 10% w/w)/NDP9 (1 nm)/Au (50 nm). In this cell architecture BPAPF serves as a hole transport material and NDP9 is a dopant. The D![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :A ratio was varied from 1:1 to 2:1 by volume and the two materials were co-evaporated at substrate temperatures between 50 °C and 100 °C. The data is summarized in Table 3 and representative J–V characteristics of solar cells comprising best performing heteropentacene 6b and C60 in a ratio of 2:1 (20 and 30 nm thickness) are shown in Fig. 6 (left). J–V curves for the whole series can be seen in ESI,‡ Fig. S2.
:C60 (2:1) at different substrate temperatures and thicknesses of 20 nm and 30 nm. The active area of the solar cells was 6.4 mm2
:A ratio was varied from 1:1 to 2:1 by volume and the two materials were co-evaporated at substrate temperatures between 50 °C and 100 °C. The data is summarized in Table 3 and representative J–V characteristics of solar cells comprising best performing heteropentacene 6b and C60 in a ratio of 2:1 (20 and 30 nm thickness) are shown in Fig. 6 (left). J–V curves for the whole series can be seen in ESI,‡ Fig. S2.
:C60 (2:1) at different substrate temperatures and thicknesses of 20 nm and 30 nm. The active area of the solar cells was 6.4 mm2
| Donor | Substrate temp. [°C] | Film thickness [nm] | J SC [mA cm−2] | V OC [V] | Saturationa | FF | PCE [%] | EQE @ 600 nm |
|---|---|---|---|---|---|---|---|---|
| a Defined as J(−1 V)/JSC. b Strong crystallization. | ||||||||
| 6a(Me) | 50 | 20 | 4.9 | 0.83 | 1.64 | 0.32 | 1.30b | 0.37 |
| 50 | 30 | 4.6 | 0.84 | 1.61 | 0.33 | 1.28b | 0.32 | |
| 6b (Et) | 50 | 20 | 10.0 | 0.89 | 1.07 | 0.67 | 5.96 | 0.63 |
| 50 | 30 | 9.5 | 0.88 | 1.08 | 0.59 | 4.93 | 0.58 | |
| 6c(Pr) | 90 | 20 | 10.4 | 0.85 | 1.05 | 0.60 | 5.30 | 0.61 |
| 90 | 30 | 11.0 | 0.85 | 1.05 | 0.59 | 5.52 | 0.64 | |
| 6d(Bu) | 80 | 20 | 9.3 | 0.93 | 1.07 | 0.68 | 5.88 | 0.63 |
| 80 | 30 | 9.0 | 0.92 | 1.08 | 0.67 | 5.55 | 0.59 | |
| 6e(Pen) | 80 | 20 | 9.6 | 0.98 | 1.10 | 0.51 | 4.48 | 0.68 |
| 80 | 30 | 9.3 | 0.97 | 1.19 | 0.45 | 4.06 | 0.65 | |
| 6f(Hex) | 90 | 20 | 9.9 | 0.97 | 1.09 | 0.52 | 4.99 | 0.62 |
| 90 | 30 | 10.6 | 0.97 | 1.13 | 0.44 | 4.52 | 0.68 | |
 | ||
| Fig. 6
J–V characteristics of heteropentacene 6b:C60 (2:1) in m–i–p bulk-heterojunction solar cells with an active layer thickness of 20 nm and 30 nm as example (left) and power conversion efficiency (PCE) versus alkyl side chain length (black line 20 nm thickness, red line 30 nm) of all heteropentacenes 6b–f:C60 (2:1) (right). | ||
Before discussing the photovoltaic data and evaluating the influence of the alkyl side chains it should be noted that the methyl derivative 6a showed a high tendency to crystallize upon evaporation and formation of the bulk-heterojunction. As a consequence, and indicated by the low fill factor of around 30%, the high saturation ≫1, and the reduced open circuit voltage (VOC), we assume that the crystallites and nanophases of this donor were too large and led to unexpected and comparably very low PCEs around 1.3%. Therefore, we exclude the data of the methyl derivative 6a from the comparative discussion. Most of the solar cells of DCV-substituted heteropentacenes 6b–f showed relatively high VOC around 0.9 V which is typical for DCV-substituted oligothiophenes and originates from their low-lying HOMOs (vide supra).4,21 Voltages below 0.9 V are a sign of beginning crystallization, which is especially visible when oligomers with short side chains (Pr, Et, Me) are heated. This is the reason, why we chose higher substrate temperatures for derivatives 6c–f. The thickness of the photoactive layer did not very much influence the short-circuit current density JSC of the cells which in all cases was between 9.0 and 11.0 mA cm−2, but differences can be seen for fill factor FF and efficiencies, which are higher for the thinner 20 nm (FF 51–68%; PCE 4.48–5.96%) compared to the 30 nm layers (FF 44–59%; PCE 4.06–5.55%). We attribute this effect to decreasing molecular order and transport ability in the thicker layers. In total, heteropentacene 6b showed close to 6% efficiency, which is the best performance of all derivatives measured under directly comparable conditions.
The development of solar cell parameters in dependence of the side chain variation should give insight into the effect of the side chain length on morphological changes. In this respect, we firstly note that derivatives 6e(Pen) and 6f(Hex) with longer alkyl chains show significantly lower fill factors (44–51%) than the others 6b–d (59–68%). This points to an impaired transport in the active mixed layer of the heteroacenes and C60 possibly due to a higher intermolecular distance of donor molecules in the bulk. In Fig. 6 (right) the evolution of the PCE vs. chain length for derivatives 6b–f is plotted for both layer thicknesses. In particular, devices with 20 nm layer thickness of the photoactive bulk-heterojunction showed an obvious odd–even effect with higher values for the even-numbered alkyl chains in 6b(Et), 6d(Bu), and 6f(Hex). The 30 nm thick layers exhibited the same trend, whereby the difference between the 6c(Pr) and 6d(Bu) is only small. For the fill factors the same odd–even effect is noticed with the exception of hexyl derivative 6f. In contrast to these findings, corresponding DCV-substituted S,N-heterohexacenes showed an odd–even effect in the opposite direction: odd-numbered derivatives had the better performance, in particular pentyl side chains led to the best efficiency of 7.1%.7b
2.5 Morphological study on the photoactive layers
At that point it was clear that the differences in photovoltaic behaviour arise from the nanomorphology of the photoactive layers. Therefore, in order to rationalize the impact of the alkyl side chain length and the resulting odd–even effect of the photovoltaic parameters, we investigated surface morphology of the photoactive layers by means of tapping mode atomic force microscopy (AFM). The layers were prepared on silicon substrates with the sequence Si/C60(10 nm)/heteropentacene 6b–f:C60 (ratio 2:1, 50–90 °C substrate temperature, 30 nm thickness) in order to be as close as possible to the real device architecture. As well, in each case also the substrate temperature was adapted to the one used in the bulk-heterojunction solar cell devices.
AFM topography and phase images of the photoactive blend layers 6b–f:C60 showed at first glance striking differences: long range imaging (10 × 10 μm2 and 5 × 5 μm2, not shown) of derivatives 6c(Pr) and 6e(Pen) gave low quality images in contrast to the blends of 6b(Et), 6d(Bu), and 6f(Hex). Short range topographic images (1 × 1 μm2) of the photoactive blend layers revealed in all cases grains of 20 to 100 nm in diameter (Fig. S3, left, ESI‡). The phase images revealed a stronger phase shift of 55° to 90° for the blends containing SN5-derivatives with even-numbered side chain length 6b, 6d, and 6f compared to only ∼20° of blends comprising odd-numbered 6c and 6e (Fig. S3, right, ESI‡). A thorough analysis of phase contrast was already published for optimized blends of 6c(Pr) and 6f(Hex), in which phase separation was proven and donor and acceptor phases could be assigned.7a Accordingly, we can rationalize similar phase separation in the images of the 6b(Et), 6d(Bu), and 6f(Hex):C60 blends.22–24 On the other hand, the narrow phase shift in the case of 6c(Pr) and 6e(Pen):C60 blends is indicative of a one-component phase, either a mixed donor–acceptor or a homophase at the surface.
3. Conclusion
In summary, we have synthesized and characterized series of S,N-heteropentacenes (4–6) with varying length of the alkyl side chains attached to the nitrogens of the pentacene backbone and with varying terminal acceptor groups. In particular, DCV-substituted S,N-heteropentacenes 6 showed high thermal stability, which is necessary for the preparation of vacuum-processed bulk-heterojunction solar cells. Their high optical absorptivity and ideal frontier orbital energy levels were not affected by the length of the alkyl side chains, whereby in contrast a noticeable influence was observed for the photovoltaic parameters in bulk-heterojunction solar cells with C60 as acceptor. The efficiencies among all materials investigated were in the range of 4–6% with the best value for ethyl derivative 6b. Interestingly, an odd–even effect with superior photovoltaic performance for the SN5-derivatives with even-numbered alkyl chains was identified and rationalized from AFM surface characterization in terms of a better phase separation. These results clearly show that despite alkyl side chains do not influence electronic properties, which are highly affected by the conjugated π-system, the synthetic effort to vary them is worthwhile, because they influence the nanomorphology of the absorber system, and thus, the photovoltaic properties in quite a subtle, but not predictable way.4. Experimental section
4.1 Materials
Toluene (VWR), tetrahydrofuran (THF, VWR), diethyl ether (VWR), and dimethylformamide (DMF, Merck) were dried and purified using a MB SPS-800 (MBraun). Dichloromethane, petroleum ether, and methanol were purchased from VWR and distilled prior to use. Sodium tert-butoxide, n-hexylamine, methylammonium chloride, ethylammonium chloride, 1-propylamine, 1-pentylamine, 1,2-dichloroethane, phosphoryl chloride, sodium bicarbonate, malononitrile, piperidine, and ammonium acetate were purchased from Merck. Pd(dba)2, Pd(dppf)Cl2, and 1-butylamine were purchased from Sigma Aldrich, n-BuLi (1.6 M in n-hexane) from Acros Organics, and 1,1′-bis(diphenylphosphino)ferrocene from Frontier Scientific. ZnCl2 (VWR) was dried in high vacuum at high temperature. 3,3′,3′′,4′-Tetrabromo-2,2′:5′,2′′-terthiophene 3, 4,5-dimethyl-4,5-dithieno[2,3-d:2,3-d′]thieno[3,2-b:4,5-b′]dipyrrole 4a and 4,5-dibutyl-4,5-dithieno[2,3-d:2,3-d′]thieno[3,2-b:4,5-b′]dipyrrole 4d were prepared according to literature.18 4,5-Dipropyl-4,5-dithieno[2,3-d:2,3-d′]thieno[3,2-b:4,5-b′]dipyrrole 4c, [4,5-dipropyl-dithieno[2,3-d:2,3-d′]thieno[3,2-b:4,5-b′]dipyrrole]-2,7-dicarbaldehyde 5c, [4,5-dipropyl-dithieno[2,3-d:2,3-d′]thieno[3,2-b:4,5-b′]dipyrrole]-2,7-bis[methan-1-yl-1-yliden]dimalononitrile 6c, 4,5-dihexyl-4,5-dithieno[2,3-d:2,3-d′]thieno[3,2-b:4,5-b′]dipyrrole 4f, [4,5-dihexyl-dithieno[2,3-d:2,3-d′]thieno[3,2-b:4,5-b′]dipyrrole]-2,7-dicarbaldehyde 5f, and [4,5-dihexyl-dithieno[2,3-d:2,3-d′]thieno[3,2-b:4,5-b′]dipyrrole]-2,7-bis[methan-1-yl-1-yliden]dimalononitrile 6f were prepared according to a previous publication.7a4.2 Instrumentation
NMR spectra were recorded on a Bruker AMX 500 (1H NMR: 500 MHz, 13C NMR: 125 MHz) or a Bruker Avance 400 (1H NMR: 400 MHz, 13C NMR: 100 MHz) at 293 K if not otherwise noted. Chemical shift values (δ) are given in parts per million (ppm) using residual solvent protons (1H NMR: δH 7.26 for CDCl3, δH 5.32 for CD2Cl2, δH 5.93 for C2D2Cl4; 13C NMR: δC 77.20 for CDCl3, δC 54.00 for CD2Cl2, δC 74.20 for C2D2Cl4) as an internal standard. The splitting patterns are designated as follows: s (singlet), d (doublet), t (triplet) and m (multiplet). The assignments are Th-α-H (thiophene protons in α-position), Th-β-H (thiophene protons in β-position), and DCVH (dicyanovinylene proton). Elemental analyses were performed on an Elementar Vario EL. Melting points were determined using a Mettler Toledo DSC 823. Thin layer chromatography was carried out on aluminum plates, pre-coated with silica gel, Merck Si60 F254. Preparative column chromatography was performed on glass columns packed with silica gel, Merck Silica 60, particle size 40–63 μm. High-resolution MALDI mass spectra (HRMS) were recorded on a MS Bruker Reflex 2 (Bruker Daltonik GmbH, Bremen, Germany), MALDI-TOF mass spectra on a Bruker Daltonics Reflex III, using trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) as a matrix. Chemical ionisation (CI) mass spectra were measured on a Finnigan MAT SSQ-7000. UV-vis absorption spectra were recorded on a Perkin Elmer Lambda 19 spectrometer. Cyclic voltammetry experiments were performed using a computer-controlled Autolab PGSTAT30 potentiostat in a three-electrode single-compartment cell with a platinum working electrode, a platinum wire counter electrode, and a Ag/AgCl reference electrode. All potentials were internally referenced to the ferrocene/ferricenium couple. Surface images of photoactive layers were recorded with the help of an atomic force microscope (MultiMode V AFM, Bruker). The microscope was operated in air at room temperature in the tapping mode using commercial silicon tips with a resonance frequency of 200–400 kHz and spring constants of ≈50 N m−1. The images were analyzed with the help of the WSxM software.254.3 Synthesis
:1) to give ethylated SN5 4b (182 mg, 0.52 mmol, 52%) as a beige solid. Mp 221 °C; 1H NMR (400 MHz, CDCl3): δ 7.10 (d, J = 5.3 Hz, 2H, Th-α-H), 7.02 (d, J = 5.3 Hz, 2H, Th-β-H), 4.47 (q, J = 7.2 Hz, 4H, N-CH2), 1.53 (t, J = 7.2 Hz, 6H, –CH3); 13C NMR (100 MHz, C2D2Cl4): δ 143.7, 129.9, 122.5, 116.7, 115.7, 111.3, 43.8, 16.6; MS (CI m/z): 330 [M+, 100%]. Anal. calcd for C16H14N2S3: C, 58.15; H, 4.27; N, 8.48; S 29.10; found: C, 58.14; H, 4.34; N, 8.32; S, 28.86.
:3) and recrystallized from petroleum ether/dichloromethane to give pentylated SN5 4e (215 mg, 0.53 mmol, 53%) as a light yellow solid. Mp 115 °C; 1H NMR (400 MHz, CDCl3): δ 7.09 (d, J = 5.2 Hz, 2H, Th-α-H), 7.00 (d, J = 5.1 Hz, 2H, Th-β-H), 4.36 (t, J = 7.6 Hz, 4H, N-CH2), 1.90 (m, 4H, N-CH2-CH2), 1.38–1.31 (m, 8H, 2(–CH2–)), 0.88 (t, J = 6.8 Hz, 6H, –CH3); 13C NMR (100 MHz, CDCl3): δ 144.3, 130.2, 122.1, 116.8, 116.4, 111.3, 49.1, 31.3, 29.3, 22.6, 14.1; MS (CI m/z): 414 [M+]. Anal. calcd for C22H26N2S3: C, 63.73; H, 6.32; N, 6.76; S, 23.20; found: C, 63.88; H, 6.30; N, 6.63; S, 22.98.
:1) affording dialdehyde 5d as an orange solid (115 mg, 0.26 mmol, 55%). Mp 284 °C; 1H NMR (400 MHz, CDCl3): δ 9.88 (s, 2H, CHO), 7.65 (s, 2H, Th-β-H), 4.42 (t, J = 7.5 Hz, 4H, N-CH2), 1.96–1.85 (m, 4H, N-CH2-CH2), 1.39 (m, 4H, –CH2-CH3), 0.97 (t, J = 9.1 Hz, 6H, –CH3); 13C NMR (125 MHz, C2D2Cl4, 363 K): δ 182.4, 145.2, 141.4, 133.7, 125.0, 120.1, 118.9, 49.3, 33.5, 20.4, 13.8; HRMS (MALDI) m/z: calcd for C22H22N2O2S3, 442.08379; found, 442.08375 [M+] (δ m/m = 0.09 ppm).
:1). Pure dialdehyde 5e was isolated as an orange solid (88 mg, 0.19 mmol, 79%). Mp: 238 °C; 1H NMR (400 MHz, C2D2Cl4): δ 9.80 (s, 2H, CHO), 7.60 (s, 2H, Th-β-H), 4.33 (t, J = 7.5 Hz, 4H, N-CH2), 1.92–1.82 (m, 4H, N-CH2-CH2), 1.36–1.26 (m, 8H, 2(–CH2–)), 0.85 (t, J = 6.7 Hz, 6H, –CH3); 13C NMR (100 MHz, C2D2Cl4): δ 183.0, 144.8, 140.8, 133.3, 124.5, 120.4, 119.6, 49.3, 31.3, 29.2, 22.5, 14.1; HRMS (MALDI) m/z: calcd for C24H26N2O2S3, 470.11509; found, 470.11499 [M+] (δ m/m = 0.2 ppm).
4.4 Thin film and device fabrication
Thin films and heterojunction solar cell devices were prepared by thermal vapor deposition in ultra-high vacuum at a base pressure of 10−7 mbar. Thin films for absorption and emission measurements were prepared on quartz substrates at room temperature. Solar cells were made on tin-doped indium oxide (ITO) coated glass (Thin Film Devices, USA, sheet resistance of 30 Ω sq−1). Layer thicknesses were determined during evaporation by using quartz crystal monitors. The thin films prepared for absorption and emission measurements were 30 nm thick. Thin film absorption spectra were recorded on a Shimadzu UV-2101/3101 UV-vis spectrometer. Bulk-heterojunction solar cells were prepared layer by layer without breaking the vacuum. The layer structure of the bulk-heterojunction solar cells is as follows: ITO; 15 nm C60; 20 (30) nm blend layer of respective donors and C60 (ratio 2:1 by mass) prepared by co-evaporation deposited on heated substrate; 5 nm BPAPF; 50 nm BPAPF doped with NDP9 (purchased from Novaled AG Germany, 10 wt%); 1 nm NDP9; 50 nm gold. Each set of devices contained two devices of 20 nm and two devices of 30 nm absorber thickness.
4.5 Photovoltaic characterization
J–V and EQE measurements were carried out in a glove box with nitrogen atmosphere. J–V characteristics were measured using a source-measure unit (Keithley SMU 2400) and an AM 1.5G sun simulator (KHS Technical Lighting SC1200). The intensity was monitored with a silicon photodiode (Hamamatsu S1337), which was calibrated at Fraunhofer ISE. The mismatch between the spectrum of the sun simulator and the solar AM 1.5G spectrum was taken into account for the calculation of current density. For well-defined active solar cell areas, aperture masks (2.76 mm2) were used. Simple EQE measurements were carried out using the sun simulator in combination with color filters for monochromatic illumination. The illumination intensities were measured with a silicon reference diode (Hamamatsu S1337).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge fruitful discussions with Dr Amaresh Mishra. We thank Dr Markus Wunderlin for the measurement of a multitude of mass spectra.References
- (a) R. Meerheim, C. Körner and K. Leo, Appl. Phys. Lett., 2014, 105, 063306 CrossRef; (b) H.-S. Shim, C.-K. Moon, J. Kim, C.-K. Wang, B. Sim, F. Lin, K.-T. Wong, Y. Seo and J.-J. Kim, ACS Appl. Mater. Interfaces, 2016, 8, 1214–1219 CrossRef CAS PubMed; (c) A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS PubMed.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148–7151 CrossRef CAS PubMed.
- D. Deng, Y. Zhang, J. Zhang, Z. Wang, L. Zhu, J. Fang, B. Xia, Z. Wang, K. Lu, W. Ma and Z. Wei, Nat. Commun., 2016, 7, 1–9 Search PubMed.
- K. Schulze, C. Uhrich, R. Schüppel, K. Leo, M. Pfeiffer, E. Brier, E. Reinold and P. Bäuerle, Adv. Mater., 2006, 18, 2872–2875 CrossRef CAS.
- Heliatek GmbH, 2016. “Heliatek sets new Organic Photovoltaic world record efficiency of 13.2%”, press release, 8(th) of February, 2016.
- B. Maennig, J. Drechsel, D. Gebeyehu, P. Simon, F. Kozlowski, A. Werner, F. Li, S. Grundmann, S. Sonntag and M. Koch, Appl. Phys. A: Mater. Sci. Process., 2004, 79, 1–14 CrossRef CAS.
- (a) A. Mishra, D. Popovic, A. Vogt, H. Kast, T. Leitner, K. Walzer, M. Pfeiffer, E. Mena-Osteritz and P. Bäuerle, Adv. Mater., 2014, 26, 7217–7223 CrossRef CAS PubMed; (b) C. Wetzel, A. Mishra, E. Mena-Osteritz, K. Walzer, M. Pfeiffer and P. Bäuerle, J. Mater. Chem. C, 2016, 4, 3715–3725 RSC; (c) C.-L. Chung, C.-Y. Chen, H.-W. Kang, H.-W. Lin, W.-L. Tsai, C.-C. Hsu and K.-T. Wong, Org. Electron., 2016, 28, 229–238 CrossRef CAS.
- (a) T. Lei, J. Y. Wang and J. Pei, Chem. Mater., 2014, 26, 594–603 CrossRef CAS; (b) J. Mei and Z. Bao, Chem. Mater., 2014, 26, 604–615 CrossRef CAS.
- A. Gadisa, W. D. Oosterbaan, K. Vandewal, J. C. Bolsée, S. Bertho, J. D’Haen, L. Lutsen, D. Vanderzande and J. V. Manca, Adv. Funct. Mater., 2009, 19, 3300–3306 CrossRef CAS.
- L. Yang, H. Zhou and W. You, J. Phys. Chem. C, 2010, 114, 16793–16800 CAS.
- J. P. Sun, A. D. Hendsbee, A. J. Dobson, G. C. Welch and I. G. Hill, Org. Electron., 2016, 36, 151–157 CrossRef CAS.
- I. Ata, S. B. Dkhil, M. Pfannmöller, S. Bals, D. Duché, J.-J. Simon, T. Koganezawa, N. Yoshimoto, C. Videlot-Ackermann, O. Margeat, J. Ackermann and P. Bäuerle, Org. Chem. Front., 2017, 4, 1561–1573 RSC.
- C. Wetzel, A. Mishra, E. Mena-Osteritz, A. Liess, M. Stolte, F. Würthner and P. Bäuerle, Org. Lett., 2014, 16, 362–365 CrossRef CAS PubMed.
- H. Kast, A. Mishra, G. L. Schulz, M. Urdanpilleta, E. Mena-Osteritz and P. Bäuerle, Adv. Funct. Mater., 2015, 25, 3414–3424 CrossRef CAS.
- (a) P. Qin, H. Kast, M. K. Nazeeruddin, S. M. Zakeeruddin, A. Mishra, P. Bäuerle and M. Grätzel, Energy Environ. Sci., 2014, 7, 2981–2985 RSC; (b) C. Steck, M. Franckevičius, S. M. Zakeeruddin, A. Mishra, P. Bäuerle and M. Grätzel, J. Mater. Chem. A, 2015, 3, 17738–17746 RSC; (c) D. Bi, A. Mishra, P. Gao, M. Franckevičius, C. Steck, S. M. Zakeeruddin, M. K. Nazeeruddin, P. Bäuerle, M. Grätzel and A. Hagfeldt, ChemSusChem, 2016, 9, 433–438 CrossRef CAS PubMed.
- K. Mitsudo, S. Shimohara, J. Mizoguchi, H. Mandai and S. Suga, Org. Lett., 2012, 14, 2702–2705 CrossRef CAS PubMed.
- M. Curtis, J. I. Nanos, H. Moon and W. Shang Jahng, J. Am. Chem. Soc., 2007, 84, 15072–15084 CrossRef PubMed.
- R. Fitzner, E. Mena-Osteritz, A. Mishra, G. Schulz, E. Reinold, M. Weil, C. Körner, H. Ziehlke, C. Elschner, K. Leo, M. Riede, M. Pfeiffer, C. Uhrich and P. Bäuerle, J. Am. Chem. Soc., 2012, 134, 11064–11067 CrossRef CAS PubMed.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367–2371 CrossRef CAS PubMed.
- Q. Xie, F. Arias and L. Echegoyen, J. Am. Chem. Soc., 1993, 115, 9818–9819 CrossRef CAS.
- R. Fitzner, E. Reinold, A. Mishra, E. Mena-Osteritz, P. Bäuerle, H. Ziehlke, C. Körner, K. Leo, M. Riede and M. Weil, Adv. Funct. Mater., 2011, 21, 897–910 CrossRef CAS.
- V. Shrotriya, Y. Yao, G. Li and Y. Yang, Appl. Phys. Lett., 2006, 89, 063505 CrossRef.
- M. Weidelener, C. D. Wessendorf, J. Hanisch, E. Ahlswede, G. Götz, M. Lindén, G. Schulz, E. Mena-Osteritz, A. Mishra and P. Bäuerle, Chem. Commun., 2013, 49, 10865–10867 RSC.
- L. Ding, J. Cao, Q. Liao, X. Du, J. Chen, Q. Zuo and Z. Xiao, Energy Environ. Sci., 2013, 6, 3224–3228 Search PubMed.
- I. Horcas, R. Fernández, J. M. Gómez-Rodríguez, J. Colchero, J. Gómez-Herrero and A. M. Baro, Rev. Sci. Instrum., 2007, 78, 013705 CrossRef CAS PubMed.
Footnotes |
| † This article is invited to a themed collection associated with the “13th International Symposium on Functional π-Electron Systems”, Guest Editors: He Yan, Ben Zhong Tang, The Hong Kong University of Science and Technology. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qm00542c |
| This journal is © the Partner Organisations 2018 |