
Poly(lactic acid)/poly(ethylene glycol) stereocomplexed physical hydrogels showing thermally-induced gel–sol–gel multiple phase transitions†
Hailiang
Mao
a,
Chen
Wang
*b,
Xiaohua
Chang
a,
Heqing
Cao
a,
Guorong
Shan
 a,
Yongzhong
Bao
a and
Pengju
Pan
*a
a,
Yongzhong
Bao
a and
Pengju
Pan
*a
aState Key Laboratory of Chemical Engineering, College of Chemical and Biological Engineering, Zhejiang University, 38 Zheda Road, Hangzhou 310027, China. E-mail: panpengju@zju.edu.cn
bKey Laboratory of Auxiliary Chemistry & Technology for Chemical Industry, Ministry of Education, Shaanxi University of Science & Technology, Xi'an 710021, China. E-mail: wangchenhg@sust.edu.cn
First published on 12th December 2017
Abstract
Stereocomplexation of complementary chiral blocks has been a feasible way to prepare thermoresponsive physical gels (or thermogels). Understanding the thermoresponsive phase behavior of stereocomplexable amphiphilic copolymers in water is essential to prepare thermogels with tunable physical properties. Herein, we use an enantiomeric mixture of the poly(D-lactic acid) (PDLA)/poly(ethylene glycol) (PEG) diblock copolymer and the poly(L-lactic acid) (PLLA)/PEG triblock copolymer as a stereocomplexable system and report a novel thermogel that exhibits unique gel–sol–gel multiple transitions upon heating. The thermally-induced phase transition of the PEG–PDLA/PLLA–PEG–PLLA thermogel was governed by the copolymer composition and the stereocomplexation of PLLA/PDLA segments. The molecular motion of PEG segments enhances during the gel–sol transition but slows down in the subsequent sol–gel transition. The gel–sol and sol–gel transitions that occurred at low and high temperatures are driven by the increased mobility of PEG segments and micelles and the promoted PLLA/PDLA stereocomplexation, respectively. PEG–PDLA/PLLA–PEG–PLLA gels can be used to capsulate and release a hydrophobic drug; the drug release rate increases as the content of hydrophilic blocks increases.
 Pengju Pan | Pengju Pan obtained his bachelor's degree from Zhengzhou University in 2002 and master's degree from Zhejiang University in 2005. He received his PhD from the Tokyo Institute of Technology in 2009. He then joined the Bioengineering Laboratory of RIKEN (Japan) as a postdoctoral researcher (2009–2011). He started his independent career at Zhejiang University in 2011. He has received the Chinese Government Award for Outstanding Self-Financed Students Abroad and the National Science Fund for Excellent Young Scholars, China. His research interests are the condensed and soft matter structures of polymers, with a focus on polymer crystallization, hierarchically assembled structures and the properties of crystallizable polymers. |
Introduction
Amphiphilic block copolymers can self-assemble into core–shell micelles in aqueous solution, because of the microphase separation between the hydrophobic and hydrophilic blocks. The stability of copolymer micelles is usually sensitive to external stimuli such as temperature, pH, and ionic strength.1 These copolymer micelles can undergo solution (sol)-to-gel transition and form physical gels when they are destabilized and aggregated under strong intermicellar interactions. A feasible stimulus to tune the sol–gel transition and physical gelation of copolymer micelles is temperature; this provides a simple yet effective way to prepare thermoresponsive physical gels (or thermogels).2 Such thermogels have shown tremendous potential applications in controlled drug delivery and tissue engineering.3–5 Due to their good biodegradability and biocompatibility, aliphatic polyesters such as poly(lactic-co-glycolic acid) (PLGA), poly(lactic acid) (PLA), and poly(ε-caprolactone) (PCL) have been widely used as the hydrophobic building blocks of amphiphilic copolymers to prepare thermoresponsive gels.4–6Depending on their chemical structure and composition, amphiphilic copolymers are able to show diversified thermally-induced phase transitions in solutions or gels. Understanding the phase behavior is of fundamental importance for the preparation of thermoresponsive gels with the desired physical properties and functions. It has been reported that amphiphilic copolymers can undergo different types of phase transitions (including sol–gel–sol, sol–gel, and gel–sol transitions) upon heating. First, triblock copolymers such as poly(ethylene glycol)–poly(propylene glycol)–poly(ethylene glycol) (PEG–PPG–PEG),7,8 PLGA–PEG–PLGA,9–11 PEG–PLGA–PEG,12 PCL–PEG–PCL,13 PEG–PCL–PEG,14 and poly(ε-caprolactone-co-lactic acid)–PEG–poly(ε-caprolactone-co-lactic acid) (PCLA–PEG–PCLA),15,16 and PEG/PLLA17 and PEG/PCL multiblock copolymers18 exhibit multiple sol–gel–sol transitions upon heating. Such copolymers change from flowable solutions to solid-like gels in the initial heating because of micellar aggregation, while the gels turn into flowable solutions upon further heating. Such phase transitions are usually reversible; the solid-like gels return to flowable solutions upon subsequent cooling. Second, some amphiphilic copolymers such as PEG–poly(L-lactic acid) (PEG–PLLA),19–21 PEG–PCL,21 PEG–PLGA,21,22 poly(D,L-lactic acid)–PEG–poly(D,L-lactic acid) (PDLLA–PEG–PDLLA), and PLLA–PEG–PLLA23 exhibit sol–gel–sol transitions in certain concentration ranges. These copolymers are solid-like gels at a low temperature but change into flowable solutions upon heating. It has been proposed that the thermally-induced gel–sol transition originates from the decrease in the occupied volume of micelles.24
The other interesting thermogel system is based on the stereocomplexable amphiphilic copolymers (or copolymer mixtures).25–34 PLLA and poly(D-lactic acid) (PDLA), both of which are biobased, biodegradable, and biocompatible, are a widely-used enantiomeric pair to prepare stereocomplexed thermogels.35 The enantiomerically-mixed copolymer solutions of PLLA–PEG–PLLA and PDLA–PEG–PDLA undergo a sol–gel transition upon heating, because of stereocomplexation-induced intermicellar crosslinking.29 Different from PLGA–PEG–PLGA thermogels, stereocomplexed thermogels are stable and cannot return to flowable solutions at high temperature.25,29 Besides, it has been reported that the PEG–PLLA and PEG–PDLA/PEG–PLLA mixture solely exhibits a gel–sol transition upon heating.28 Therefore, the PEG–PDLA/PLLA–PEG–PLLA mixture may show more diversified phase behavior than the well-studied PDLA–PEG–PDLA/PLLA–PEG–PLLA system. Using the stereocomplexation of PLLA and PDLA blocks, we have found that the solutions of PLA/PEG diblock and triblock copolymers having opposite PLA chirality form physical gels immediately after mixing at room temperature.36
For copolymeric gels and concentrated solutions consisting of stereocomplexable hydrophobic segments, the stereocomplexation of hydrophobic segments (e.g., PLLA and PDLA) would be promoted upon heating; this promotes the formation of stereocomplexed physical networks between the micelles and thus facilitates the thermally-induced sol–gel transition. On the other hand, physical gels made from amphiphilic copolymers tend to transform into flowable solutions at a high temperature, because of the increased mobility of hydrophilic segments and micelles or the decrease in the occupied volume of micelles.24 Due to the synergistic effects of the stereocomplexation of hydrophobic blocks and the mobility of micelles, the stereocomplexable copolymers (or copolymer mixtures) may show diversified and unique phase transition behavior in the heating process. Control over the phase transition of such copolymers can open a new way to prepare thermogels with the desired phase and rheological behavior.
In this article, we select an enantiomeric copolymer mixture of PEG–PDLA and PLLA–PEG–PLLA as a stereocomplexable and gellable system, and investigate the thermoresponsive phase transitions of PEG–PDLA/PLLA–PEG–PLLA enantiomeric mixtures in gels or concentrated solutions. The effects of copolymer composition and the mixing ratio on the thermoresponsive phase transition behavior were studied. For the first time, we have found an unusual, thermally-induced gel–sol–gel multiple phase transition phenomenon in the stereocomplexed thermogel through elaborately tuning the compositions of the PEG–PDLA and PLLA–PEG–PLLA copolymers used. The microstructural changes in the multiple gel–sol–gel transition of the thermogel were examined by temperature-variable 1H nuclear magnetic resonance (NMR), synchrotron-radiation wide-angle X-ray diffraction (WAXD), and small-angle X-ray scattering (SAXS). The underlying mechanism for the multiple phase transitions of the stereocomplexed thermogel was also proposed and discussed.
Experimental
Materials
Monomethoxy PEG (Mn = 1.0 kg mol−1), dihydroxyl-terminated PEGs (Mn = 2.0, 4.0, 6.0, and 10.0 kg mol−1), and tin(II) 2-ethylhexanoate [Sn(Oct)2] were purchased from Sigma-Aldrich. D- and L-lactides were obtained from Purac Co. (Gorinchem, the Netherlands), which were recrystallized from ethyl acetate before use. Toluene was dried with sodium and distilled after refluxing for 48 h. Doxorubicin hydrochloride (DOX·HCl) was purchased from Beijing Huafeng United Technology Co.Synthesis of PEG–PDLA and PLLA–PEG–PLLA block copolymers
PEG–PDLA and PLLA–PEG–PLLA block copolymers with different compositions were prepared via the ring-opening polymerization (ROP) of lactides using PEG as the macroinitiator, according to a published method.36 The successful synthesis of PLA/PEG block copolymers with controlled molecular weights and compositions was confirmed by 1H NMR and gel permeation chromatography (GPC), as shown in Fig. S1 and S2 (see the ESI†). Table 1 shows the molecular weights and polydispersities of the synthesized PLA/PEG block copolymers. PEG–PDLA and PLLA–PEG–PLLA copolymers are referred to as ExDy and LyExLy, where E, L, and D represent the PEG, PLLA, and PDLA blocks, respectively. The subscripts x and y denote the degrees of polymerization (DP) of the corresponding blocks derived from 1H NMR.| Sample | M n of each block (kg mol−1) | M n,NMR (kg mol−1) | M n,GPC (kg mol−1) | PDIb |
|---|---|---|---|---|
| a M n,NMR derived from 1H NMR. b M n,GPC and the polydispersity index (PDI) of molecular weight measured using GPC. | ||||
| E23D10 | 1.0–0.72 | 1.70 | 2.20 | 1.15 |
| E23D12 | 1.0–0.86 | 1.85 | 2.40 | 1.17 |
| E23D14 | 1.0–1.03 | 2.03 | 2.65 | 1.22 |
| E23D16 | 1.0–1.15 | 2.15 | 2.75 | 1.25 |
| L14E45L14 | 0.99–2.0–0.99 | 3.98 | 6.05 | 1.10 |
| L14E90L14 | 1.00–4.0–1.00 | 6.00 | 9.40 | 1.04 |
| L14E136L14 | 1.01–6.0–1.01 | 8.03 | 12.6 | 1.04 |
| L14E227L14 | 0.97–10.0–0.97 | 12.0 | 19.0 | 1.03 |
| L10E90L10 | 0.72–4.0–0.72 | 5.45 | 8.55 | 1.03 |
| L12E90L12 | 0.86–4.0–0.86 | 5.72 | 9.05 | 1.04 |
| L16E90L16 | 1.16–4.0–1.16 | 6.33 | 9.94 | 1.04 |
Preparation of hydrogels and analysis of thermoresponsive phase transition
PEG–PDLA and PLLA–PEG–PLLA copolymers were separately dissolved in deionized water under vigorous stirring at 20 °C. The solutions of PEG–PDLA and PLLA–PEG–PLLA were cooled to 10 °C and mixed at predetermined mass ratios to obtain an enantiomeric mixture. The enantiomeric mixture (1.0 g) was loaded into a vial (5 mL) and heated from 10 to 70 °C with a 2 °C interval in a water bath. The enantiomeric mixtures were equilibrated at each temperature interval for 10 min and then their physical states (gel or solution) were evaluated via the test tube inverting method.25,29 The enantiomeric mixture was recognized as a gel if it did not flow within 10 s; otherwise, it was regarded as a solution.29 The DP of PLLA and PDLA blocks were kept the same and the DP of the PEG block in PEG–PDLA was fixed at 23 in all the enantiomeric mixtures. For simplicity, we denote the DP of the PLLA or PDLA block as DPPLA and the DP of PEG in PLLA–PEG–PLLA as DPPEG,tri.In vitro drug release
Dehydrochloride DOX (400 μg) was dissolved in 0.5 g of PEG–PDLA solution (15 wt%) and stirred at 25 °C for 12 h. It was then mixed with 0.5 g of PLLA–PEG–PLLA solution (15 wt%) in a 5 mL vial. The enantiomerically-mixed solution formed a DOX-loaded gel (DOX concentration: 0.04 wt%) after mixing at 25 °C. The vial was placed in a plastic tube (50 mL) containing 20 mL of phosphate buffer (PB, 10 mM, pH 7.4), which was then incubated at 37 °C under shaking. At selected time intervals, 15 mL of PB was removed from the plastic tube and replaced by fresh PB. The DOX concentration in PB was measured based on the UV-Vis absorbance at 485 nm.37 The release experiments were conducted in triplicate and the averaged result was used.Measurements
Results and discussion
Thermoresponsive phase behavior of PEG–PDLA/PLLA–PEG–PLLA mixtures
When the copolymer concentration (C) is less than 18 wt%, all the PEG–PDLA and PLLA–PEG–PLLA enantiopure copolymers are in the solution state before mixing. In general, the PLA/PEG amphiphilic block copolymers would form core–shell micelles with a hydrophobic PLA core and hydrophilic PEG shell in water. Dynamic light scattering (DLS) results have verified the formation of nanometer-scaled micelles for PEG–PDLA, PLLA–PEG–PLLA, and their enantiomeric mixture in the dilute aqueous solution (0.02 wt%, Fig. S3, ESI†).Fig. 1 shows the concentration-dependent phase behaviors of PEG–PDLA/PLLA–PEG–PLLA 5/5 enantiomeric mixtures with different copolymer compositions upon heating. All the enantiomeric mixtures are solutions for C < 8 wt%, but gels for C > 18 wt% in the whole temperature range investigated (10–70 °C). The enantiomeric mixtures are thermoresponsive for 8 < C < 18 wt%; they undergo diversified phase transitions (including sol–gel, gel–sol–gel, and gel–sol transitions) upon heating. The thermoresponsive phase behavior of the enantiomeric mixtures is significantly influenced by C, DPPLA, and DPPEG,tri. The mixture with small DPPLA (e.g., E23D10/L10E90L10 5/5) is always solution within the whole C (8–18 wt%) and temperature (10–70 °C) ranges investigated.
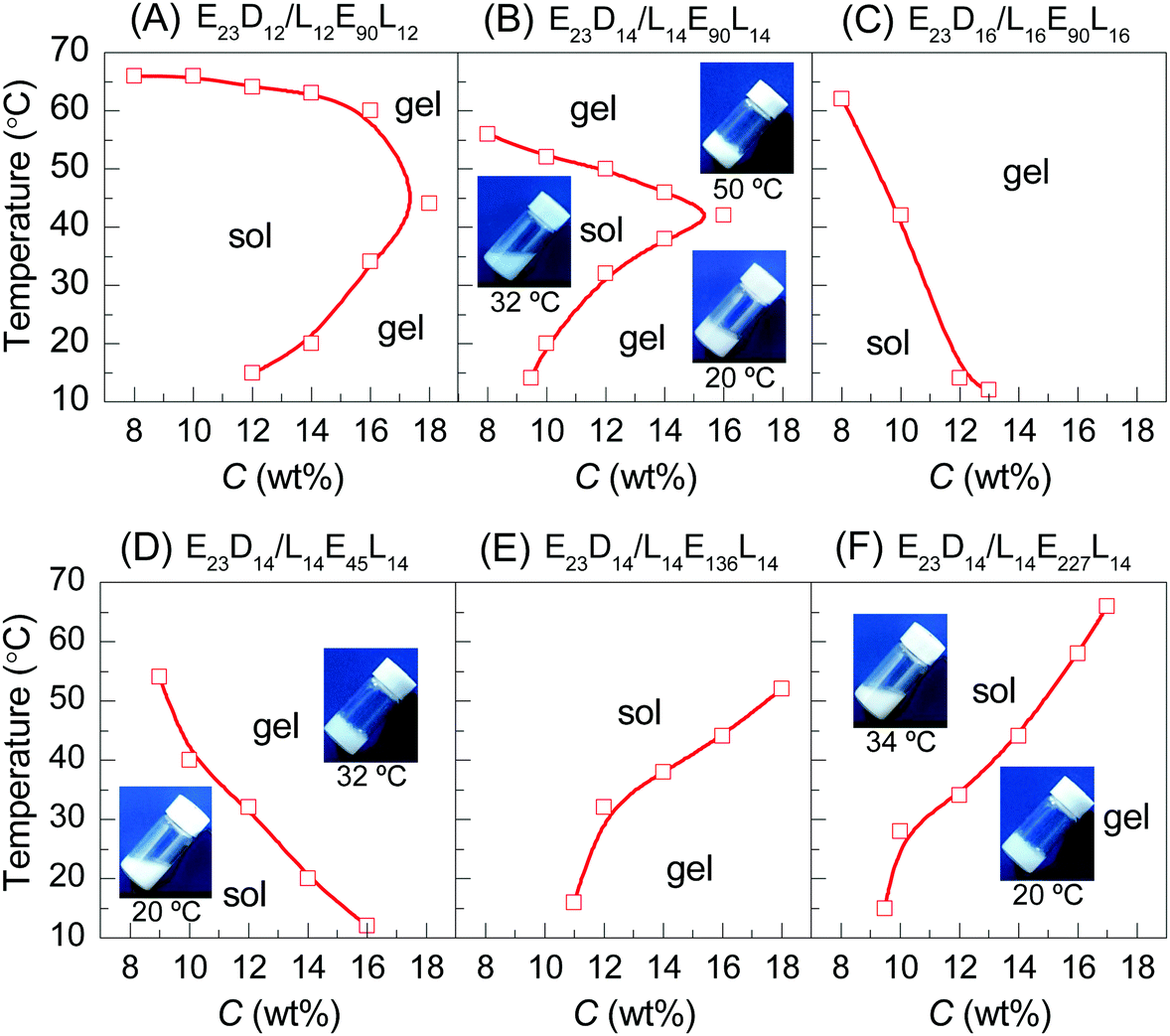 | ||
| Fig. 1 Phase diagrams of PEG–PDLA/PLLA–PEG–PLLA 5/5 mixtures at different concentrations in water: (A–C) the enantiomeric mixtures with various PEG block lengths of PEG–PDLA; (D–F) the enantiomeric mixtures with different PLLA and PDLA block lengths in PEG–PDLA and PLLA–PEG–PLLA. Digital photographs in the figures indicate the physical states of the enantiomeric mixtures (12 wt%) at different temperatures. | ||
As shown in Fig. 1A and B, the enantiomeric mixtures with medium DPPLA (e.g., E23D12/L12E90L12 5/5 and E23D14/L14E90L14 5/5) are gels at low temperature (e.g., 10 °C); they display unique gel–sol–gel multiple transitions upon heating, which is opposite to the heating-induced sol–gel–sol transition that occurred in PLGA–PEG–PLGA.9,10 The region of the solution phase becomes narrower as DPPLA increases. As shown in the inset images of Fig. 1B, both the physical gels formed at the low and high temperatures are opaque. The critical temperature of the gel–sol transition (Tg–s), located at a lower temperature, increases with C, but the critical temperature of the sol–gel transition (Ts–g) that located at a higher temperature decreases with C. As the DPPLA increases further, the enantiomeric mixture (E23D16/L16E90L16 5/5) merely exhibits a sol–gel transition upon heating at C = 8–14 wt%; the Ts–g value decreases as C increases from 8 to 14 wt% (Fig. 1C). This thermally-induced sol–gel transition is similar to that observed in a previous work.25
Interestingly, the thermally-induced gel–sol–gel transition just takes place for the enantiomeric mixtures with a suitable DPPEG,tri. As shown in Fig. 1D, the mixture with small DPPEG,tri (E23D14/L14E45L14 5/5) exhibits a sol–gel phase transition upon heating; this resembles the stereocomplexation-induced thermogelation of the PLLA–PEG–PLLA/PDLA–PEG–PDLA mixture.25,29 However, the mixtures with large DPPEG,tri (E23D14/L14E136L14 5/5, E23D14/L14E227L14 5/5) show an usual gel–sol transition upon heating, similar to the phase behavior of the in situ formed physical gels of PEG–PDLA/PLLA–PEG–PLLA mixtures.36
We emphasize that the thermally-induced phase transitions of the PEG–PDLA/PLLA–PEG–PLLA mixtures are irreversible. Upon cooling from 70 to 10 °C, the mixtures that show thermally-induced gel–sol–gel (E23D12/L12E90L12 5/5, E23D14/L14E90L14 5/5) and sol–gel transitions (E23D14/L14E45L14 5/5, E23D16/L16E90L16 5/5) are always in the gel state and do not return to the solution state. Such irreversible thermoresponsive behavior is similar to those reported for PDLA–PEG–PDLA/PLLA–PEG–PLLA stereocomplexed gels,25–27,29 which is due to the formation of stable stereocomplexed physical networks between the micelles upon heating.29 We also studied the phase behavior of a premixed system, in which PEG–PDLA and PLLA–PEG–PLLA were mixed and dissolved in water together (Fig. S4, ESI†). The mixing method has little influence on the phase behavior of the PEG–PDLA/PLLA–PEG–PLLA mixture. As shown in Fig. S4 (ESI†), the phase behavior of the premixed system is almost the same as that shown in Fig. 1, except for slight differences in Ts–g and Tg–s values.
The effect of the mixing ratio on the thermally-induced phase transition behavior of PEG–PDLA/PLLA–PEG–PLLA mixtures was further investigated, as shown in Fig. 2. The thermally-induced phase transition was just observed when the fractions of PEG–PDLA and PLLA–PEG–PLLA are similar or comparable in the mixtures. The E23D14/L14E90L14 mixture shows a thermally-induced gel–sol–gel transition within a given range of mixing ratios (7/3–3/7), while the phase behavior of the mixture with the other mixing ratios is insensitive to temperature (Fig. 2B). Similarly, the thermally-induced sol–gel or gel–sol transitions are mainly observed in the E23D14/L14E45L14 and E23D14/L14E227L14 mixtures when the PEG–PDLA/PLLA–PEG–PLLA mixing ratios are between 8/2 and 2/8 or 7/3 and 3/7. These suggest that the physical gelation of the enantiomeric mixtures originates from the stereocomplexation of PLLA and PDLA blocks, as will be demonstrated in the following sections.
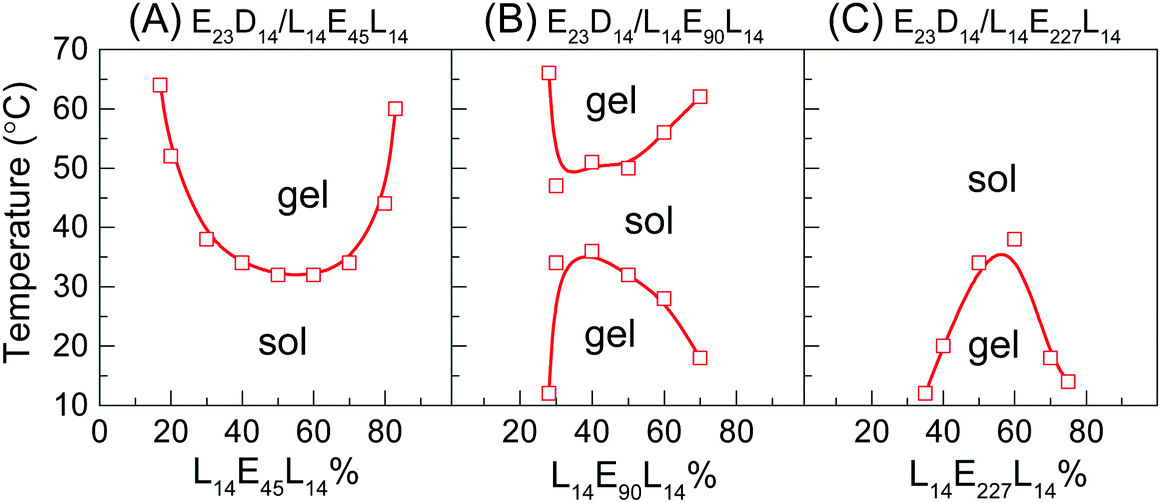 | ||
| Fig. 2 Phase diagrams of PEG–PDLA/PLLA–PEG–PLLA enantiomeric mixtures (12 wt%) with different mixing ratios. | ||
The thermoresponsive phase behavior of PEG–PDLA/PLLA–PEG–PLLA mixtures was further investigated by rheological analysis. Fig. 3 shows the temperature-dependent storage moduli (G′) and loss moduli (G′′) of the PEG–PDLA/PLLA–PEG–PLLA 5/5 mixtures with different copolymer compositions. The higher G′ relative to G′′ is characteristic of the gel state, while G′′ > G′ is a feature of the solution state. The crossover point of G′ and G′′ corresponds to the sol–gel or gel–sol transition.18 As shown in Fig. 3A, the E23D14/L14E45L14 5/5 mixture is a solution at <∼31 °C but turns into a gel at >∼31 °C. As shown in Fig. 3B, the E23D14/L14E90L14 5/5 mixture undergoes a gel–sol transition at 31 °C and then a sol–gel transition at 50 °C during heating. The G′ value of the E23D14/L14E90L14 mixture is 229 Pa at 60 °C, which is much higher than the value of 32 Pa at 10 °C; the higher modulus at higher temperature is ascribed to the improved stereocomplexation of PLLA/PDLA blocks, as elaborated in the following WAXD results. As shown in Fig. 3C, both the G′ and G′′ of the E23D14/L14E227L14 5/5 mixture decrease upon heating; this mixture shows Tg–s at 33 °C. Even though the transition temperatures measured by the rheological and test tube methods are slightly different, these rheological results can demonstrate diversified thermoresponsive phase transitions of PEG–PDLA/PLLA–PEG–PLLA mixtures. Because the thermally-induced gel–sol–gel transition is quite unique and unusual for physical gels, we focus on the E23D14/L14E90L14 5/5 thermogel and its thermoresponsive phase transition in this article.
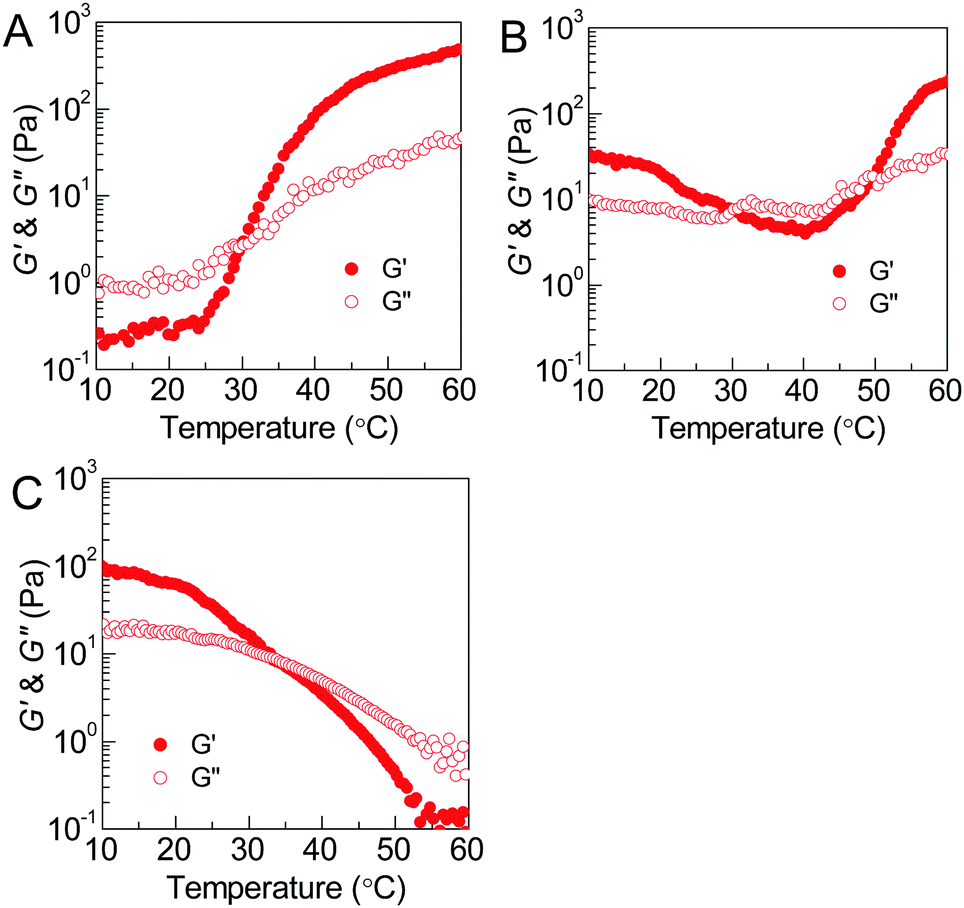 | ||
| Fig. 3 Plots of storage moduli as a function of temperature for PEG–PDLA/PLLA–PEG–PLLA 5/5 enantiomeric mixtures (12 wt%) at 1 Hz: (A) E23D14/L14E45L14; (B) E23D14/L14E90L14; and (C) E23D14/L14E227L14. | ||
Thermoresponsive phase transition analyzed by NMR
The thermoresponsive phase transition of the PEG–PDLA/PLLA–PEG–PLLA gel was examined by temperature-variable 1H NMR spectroscopy. Panel A of Fig. 4 shows the 1H NMR spectra of the E23D14/L14E90L14 5/5 thermogel measured upon heating from 20 to 60 °C. The corresponding spectra of the E23D10/L14E90L10 5/5 solution (control sample) are shown in Fig. 4B for comparison; this sample is always in the solution state and does not undergo phase transition during heating. The peaks around 1.6, 3.7, and 4.7 ppm refer to the methyl proton signals of PLLA (or PDLA), the methylene proton signals of PEG, and the solvent peak of D2O, respectively. D2O is a good solvent for PEG blocks but a poor solvent for PLLA and PDLA blocks. Compared with the 1H NMR spectra of block copolymers in CDCl3 (Fig. S1, ESI†), the methyl proton signals of PLLA or PDLA are very weak and broad, whereas the methylene proton signals of PEG are sharp and strong. This suggests that PLLA and PDLA blocks self-assemble in the hydrophobic domains (i.e., micelle cores) in the PEG–PDLA/PLLA–PEG–PLLA gel and solution.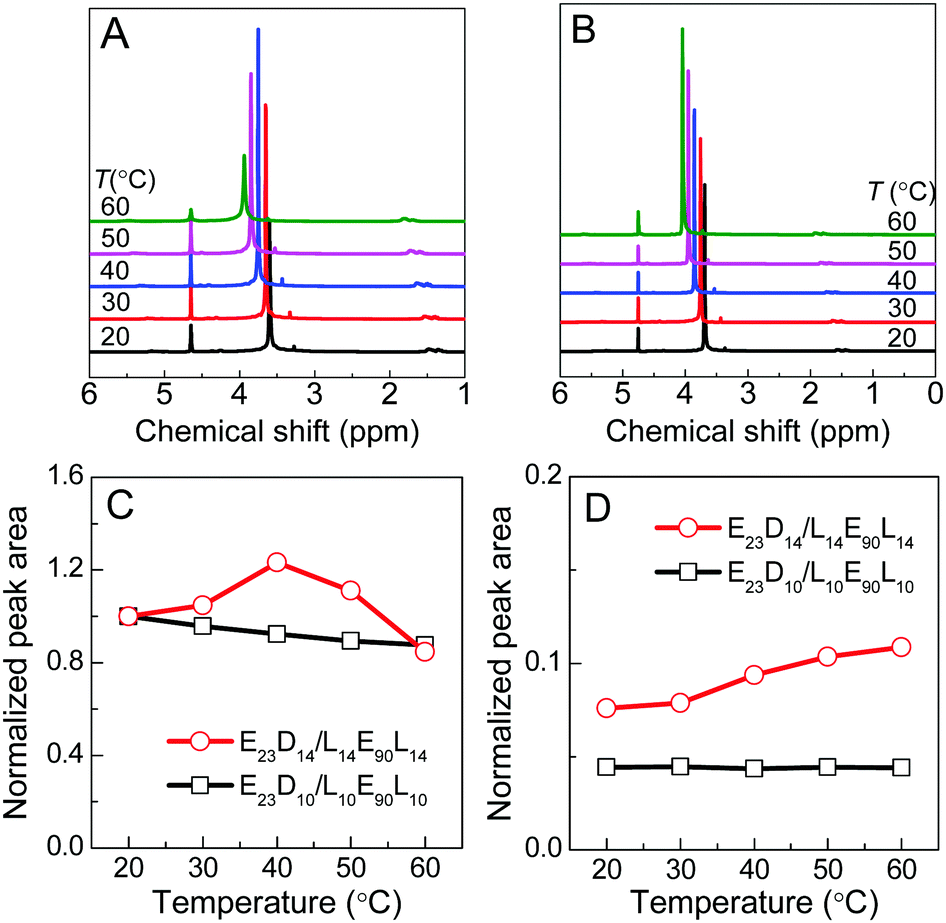 | ||
| Fig. 4 Temperature-variable 1H NMR results of the PEG–PDLA/PLLA–PEG–PLLA 5/5 gel and solution (12 wt%) in D2O: (A) NMR spectra of the E23D14/L14E90L14 5/5 gel; (B) NMR spectra of the E23D10/L10E90L10 5/5 solution (control sample); (C) normalized peak area of PEG methylene protons plotted against temperature; and (D) normalized peak area of PLLA/PDLA methyl protons plotted against temperature. The peak area of each sample was normalized by the peak area of PEG methylene protons measured at 20 °C. | ||
As shown in Fig. 4A and B, the chemical shift of the D2O signal is nearly constant, whereas the PEG and PLA signals shift downfield by ∼0.1 ppm every 10 °C during heating, indicating the decrease in the solvation of PEG and PLLA (or PDLA) segments at higher temperature. On the basis of the NMR spectra, we evaluated the thermally-induced intensity changes of PEG methylene protons and PLLA/PDLA methyl protons (as shown in Fig. 4C and D), which reflects the variation in the molecular environment for the PEG and PLLA/PDLA segments. The stronger intensity and narrower width of the NMR signal mean the freer motion and higher mobility of the involved chain segments. For the control sample (E23D10/L14E90L10 5/5) that is always in the solution state, the NMR intensity of PEG methylene protons decreases slightly and that of PLLA/PDLA methyl protons changes little upon heating (Fig. 4C and D). However, in the case of the E23D14/L14E90L14 5/5 mixture, the NMR intensity of PEG methylene protons increases upon heating from 20 to 40 °C (corresponding to the gel–sol transition) and then decreases with further heating from 40 to 60 °C (corresponding to the sol–gel transition), suggesting first the increase and then the decrease of PEG segmental mobility during heating (Fig. 4C). Therefore, it is concluded that PEG segments have higher mobility in the solution state, while they are restricted in the gel state. As shown in Fig. 4D, the NMR intensity of PLLA/PDLA methyl protons slightly increases with heating, implying the enhanced mobility of PLLA/PDLA segments at higher temperature.
Crystalline and microstructural changes in thermoresponsive phase transition
The crystalline structural evolutions of PLLA/PDLA hydrophobic segments in the PEG–PDLA/PLLA–PEG–PLLA gel were analyzed by synchrotron-radiation WAXD and SAXS. Panel A of Fig. 5 shows the temperature-variable WAXD patterns of the E23D14/L14E90L14 5/5 gel collected upon heating from 20 to 60 °C. The corresponding profiles of the E23D10/L14E90L10 5/5 solution (control sample) are shown in Fig. 5B for comparison. As shown in Fig. 5A and B, both the diffractions of PLLA/PDLA homocrystallites (HCs) and stereocomplexes (SCs) are seen in the WAXD patterns. The existence of SCs in both the gel and solution samples may indicate that chain exchange takes place upon mixing the PEG–PDLA and PLLA–PEG–PLLA solutions in the initial stage. Alternatively, SC formation may be also induced by the absorption of PEG–PDLA chains into PLLA–PEG–PLLA micelles, since the mobility of diblock copolymers is higher than that of triblock copolymers.38 | ||
| Fig. 5 Temperature-variable WAXD results of the PEG–PDLA/PLLA–PEG–PLLA 5/5 gel and solution (12 wt%) collected upon heating: (A) WAXD patterns of the E23D14/L14E90L14 5/5 gel; (B) WAXD patterns of the E23D10/L10E90L10 5/5 solution (control sample); (C) changes in SC110 and HC110/200 diffraction peak areas upon heating; and (D) changes in fSC upon heating. | ||
To quantitatively evaluate the structural changes during heating, the peak areas of SC110 and HC110/200 diffractions, reflecting the relative contents of SCs and HCs in the PLLA/PDLA hydrophobic domains, were calculated and plotted as a function of temperature in Fig. 5C. The relative fraction of SCs (fSC) in the crystalline phase of PLLA/PDLA domains was estimated by comparing the diffraction peak area of SCs with the total areas of both SC and HC diffractions [fSC = ISC/(ISC + IHC)].36,39
As shown in Fig. 5C and D, the SC110 and HC110/200 intensities and fSC of the control sample (E23D10/L10E90L10 5/5) change little during heating. However, the SC110 and HC110/200 intensities and fSC of the E23D14/L14E90L14 5/5 gel increase gradually during heating; more distinct increases are observed upon heating from 40 to 50 °C, corresponding to the sol–gel transition. These WAXD results indicate the significantly enhanced PLLA/PDLA stereocomplexation in the sol–gel transition at 40–50 °C. Stereocomplexation can promote the exchange of hydrophobic PLLA/PDLA blocks between the different micelles, leading to the formation of intermicellar physical networks and thus driving the sol–gel transition.29
Panel A of Fig. 6 shows the temperature-variable SAXS patterns of the E23D14/L14E90L14 5/5 gel upon heating from 20 to 60 °C. The corresponding SAXS profiles of the E23D10/L14E90L10 5/5 solution (control sample) are shown in Fig. 6B for comparison. The X-axis of the SAXS profile is the scattering vector q = (4π/λ)sin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ, where 2θ is the scattering angle and λ is the X-ray wavelength. In the low-q regime (q < 0.1 nm−1), there is a significant increase in intensity for all samples, following a power law (I ∝ q−m)40 with indexes (m) of 2.4–3.0. In the mid-q regime (q = 0.1–0.6 nm−1), the SAXS profile shows a broad shoulder, attributed to the correlation between copolymer micelles.41 The correlation peak indicates stronger interactions between the micelles in the gels or concentrated solutions of the enantiomeric mixtures. In the high-q regime (q > 0.6 nm−1), the SAXS profile displays a gentle power-law slope that reflects the local structure of the micelles.
θ, where 2θ is the scattering angle and λ is the X-ray wavelength. In the low-q regime (q < 0.1 nm−1), there is a significant increase in intensity for all samples, following a power law (I ∝ q−m)40 with indexes (m) of 2.4–3.0. In the mid-q regime (q = 0.1–0.6 nm−1), the SAXS profile shows a broad shoulder, attributed to the correlation between copolymer micelles.41 The correlation peak indicates stronger interactions between the micelles in the gels or concentrated solutions of the enantiomeric mixtures. In the high-q regime (q > 0.6 nm−1), the SAXS profile displays a gentle power-law slope that reflects the local structure of the micelles.
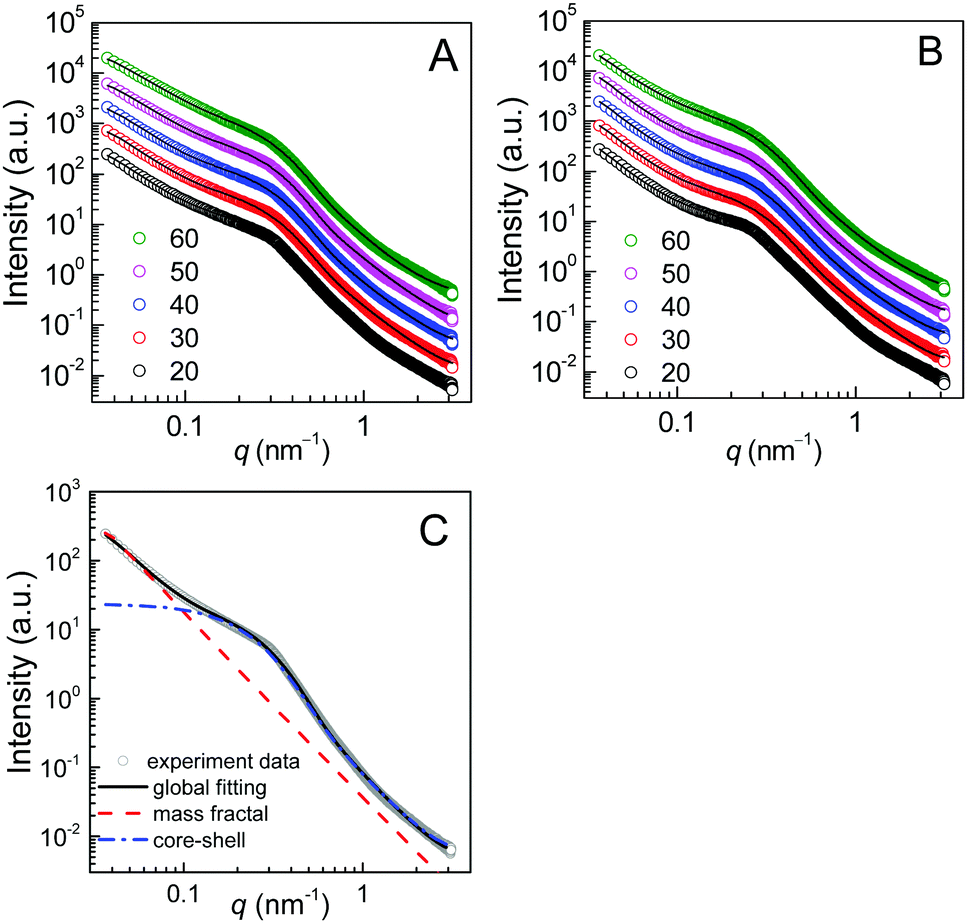 | ||
| Fig. 6 Temperature-variable SAXS results of the PEG–PDLA/PLLA–PEG–PLLA 5/5 gel and solution (12 wt%): (A) SAXS patterns of the E23D14/L14E90L14 5/5 gel; (B) SAXS patterns of the E23D10/L10E90L10 5/5 solution (control sample); and (C) model fitting by eqn (1) for the SAXS pattern of the E23D14/L14E90L14 5/5 gel collected at 20 °C. For clarity, the SAXS data collected at 30, 40, 50, and 60 °C were multiplied by 2, 4, 8, and 16 in panels A and B, respectively. | ||
Although the PEG–PDLA and PLLA–PEG–PLLA copolymers have a core–shell micelle structure in dilute solutions, the micelles interact and associate and are physically crosslinked to form larger aggregates or clusters in the gels or concentrated solutions, which would be further promoted by the stereocomplexation of PLLA/PDLA segments. To obtain detailed structural information, the core–shell form factor41,42 combined with the Teixeira model (structure factor, describing the scattering from fractal-like clusters/aggregates)43,44 was used to fit the SAXS data. This fitting model considers the scattering from a fractal structure with a primary building block of polydisperse core–shell spherical particles; it has been used to describe the scattering of PLLA–PEG–PLLA/PDLA–PEG–PDLA gels and solutions.42 The total scattering intensity I(q) can be described by the following equation:
| I(q) = nV2(Δρ)2P(q)S(q) | (1) |
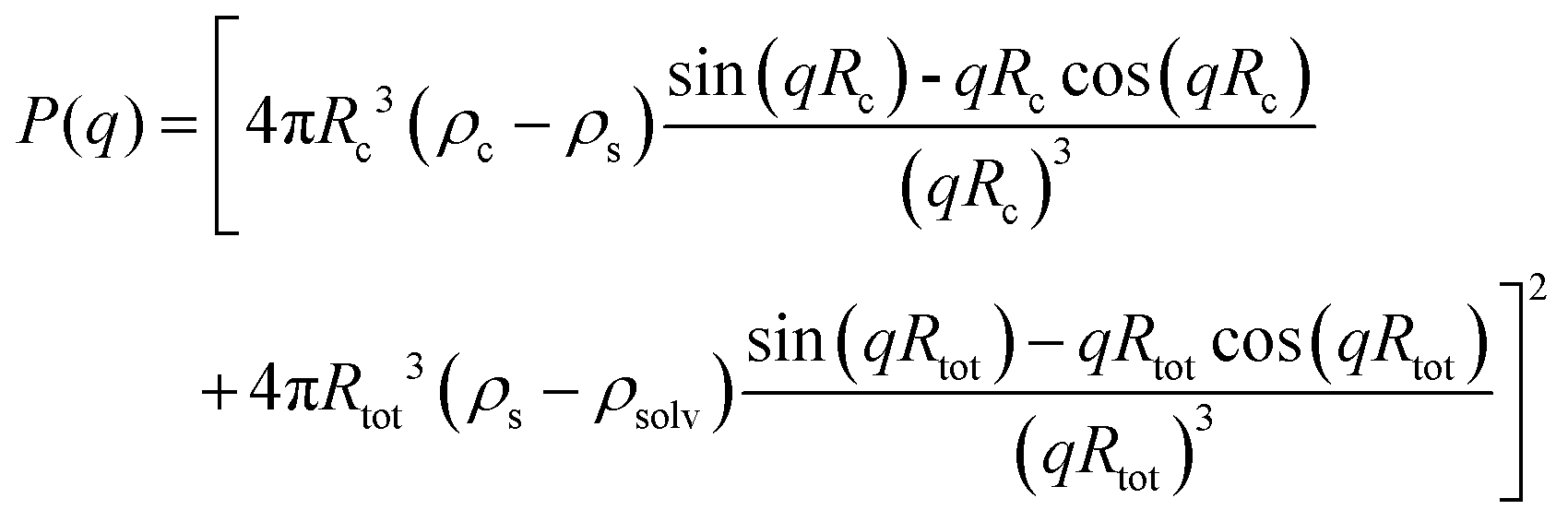 | (2) |
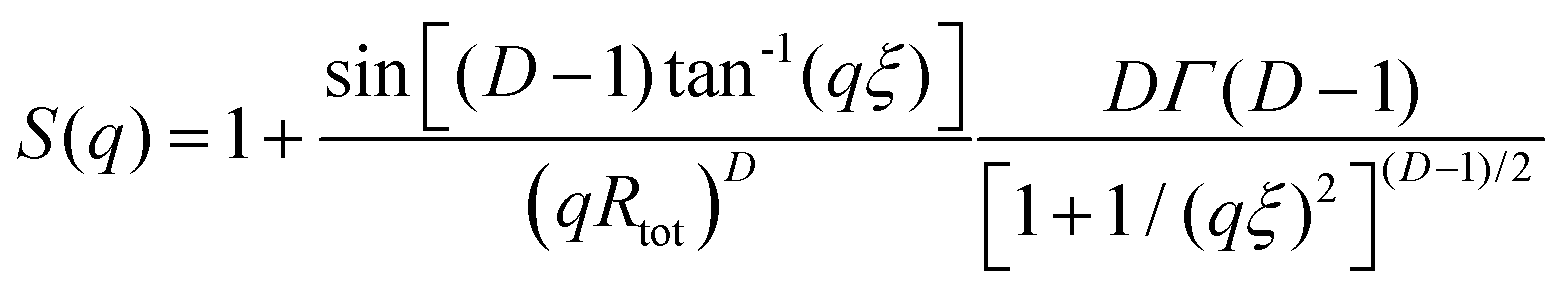 | (3) |
Fig. 6C shows the curve fitting of SAXS profiles by eqn (1). Good fits were achieved for the SAXS profiles obtained at various temperatures, indicating that the core–shell structure of the micelles is maintained in the thermally-induced phase transitions of the gel. Table 2 lists the fitted parameters including D, ξ, Rc, and shell thickness (Ls, Ls = Rtot − Rc) values for the E23D14/L14E90L14 5/5 gel and E23D10/L10E90L10 5/5 solution. As shown in Table 2, the D value of the control sample (E23D10/L10E90L10 5/5) decreases from 2.61 to 2.41 upon heating, indicating that the intermicellar packing in solution becomes looser at higher temperature. The D value of the E23D14/L14E90L14 5/5 gel changes little and maintains at 2.85–2.86 during heating. Also, the ξ values of both the E23D14/L14E90L14 5/5 gel and control sample vary little upon heating. Compared to the control sample, the Rc value of the E23D14/L14E90L14 5/5 gel gradually increases during heating; this may be due to the dehydration of PLLA/PDLA segments in the micelle cores at higher temperatures, as demonstrated by the aforementioned NMR results (Fig. 4). Because the lengths of PEG blocks are the same in the E23D14/L14E90L14 5/5 gel and control sample, the two samples show similar Ls values (1.2–1.3 nm). The Ls values of both samples are much smaller than the end-to-end distance of the PEG homopolymer (6.36 nm for PEG with a Mn of 4 kg mol−1),44 indicating that the hydrophilic PEG segments are in a highly constrained state in the gel and concentrated solution.
| T (°C) | E23D14/L14E90L14 | E23D10/L10E90L10 | ||||||
|---|---|---|---|---|---|---|---|---|
| D | ξ (nm) | R c (nm) | L s (nm) | D | ξ (nm) | R c (nm) | L s (nm) | |
| D, fractal dimension; ξ, fractal cutoff size; Rc, radius of the core; Ls, thickness of the shell. | ||||||||
| 20 | 2.86 | 23.5 | 5.6 | 1.2 | 2.61 | 20.1 | 5.2 | 1.3 |
| 30 | 2.85 | 24.5 | 5.6 | 1.2 | 2.59 | 21.0 | 5.2 | 1.3 |
| 40 | 2.85 | 24.5 | 5.7 | 1.2 | 2.54 | 21.1 | 5.2 | 1.3 |
| 50 | 2.86 | 24.7 | 5.9 | 1.3 | 2.49 | 21.1 | 5.3 | 1.3 |
| 60 | 2.86 | 24.8 | 6.0 | 1.3 | 2.41 | 21.4 | 5.3 | 1.3 |
Mechanistic discussion on thermally-induced gel–sol–gel transition
Based on the NMR, WAXD, and SAXS results, we proposed the mechanism for the thermally-induced gel–sol–gel transition of the PEG–PDLA/PLLA–PEG–PLLA thermogel (C = 12 wt%), as illustrated in Fig. 7. The PEG–PDLA copolymer forms star-shaped micelles and the PLLA–PEG–PLLA copolymer self-assembles into flower-like micelles in aqueous solution. As illustrated in Fig. 7, when the solutions of star-shaped and flower-like micelles are mixed at a low temperature (e.g., 10 °C), PEG–PDLA and PLLA–PEG–PLLA can exchange between the different micelles, which is highly promoted by the PLLA/PDLA stereocomplexation (as verified by the WAXD results in Fig. 5).36 If the two endblocks of PLLA–PEG–PLLA enter the same micelle, then stereocomplexed micelles will be formed. On the other hand, PEG–PDLA chains can absorb into PLLA–PEG–PLLA micelles to form stereocomplexed micelles with larger sizes, due to the higher mobility of the diblock copolymer;38 this is supported by the increases in micelle size upon mixing (Fig. S3, ESI†). Because the stereocomplexed micelles have a more condensed core structure and larger size than the homocrystalline micelles of PEG–PDLA or PLLA–PEG–PLLA, they may tend to aggregate in larger clusters through stronger intermicellar attractions.23 On the other hand, if the two endblocks of PLLA–PEG–PLLA enter different micelles, then stereocomplexed, intermicellar physical networks will be formed.36 Therefore, the PEG–PDLA/PLLA–PEG–PLLA enantiomeric mixture forms a physical gel at a low temperature through intermicellar physical networks or stronger intermicellar attractions. As indicated by the DLS results, the distribution of micelle size broadens drastically after mixing the dilute solutions (0.02 wt%) of PEG–PDLA and PLLA–PEG–PLLA at 20 °C (Fig. S3, ESI†). This also verifies the formation of large micelles and physical networks, and the increase in intermicellar interactions in the enantiomeric mixture.25,45,46 As illustrated in Fig. 7, when the PEG–PDLA/PLLA–PEG–PLLA thermogel is heated to a medium temperature (e.g., 30–40 °C), the mobility of the micelles increases. This improves the flowability of gel and decreases intermicellar interactions, and thus induces a gel–sol transition. However, as the enantiomerically-mixed solution is further heated (∼50 °C), the stereocomplexation of PLLA/PDLA segments is highly promoted (as demonstrated by the WAXD results in Fig. 5), leading to the formation of more condensed physical networks and thus causing the second physical gelation (i.e., sol–gel transition). Because the stereocomplexed networks are thermodynamically stable and do not destroy with cooling, the thermoresponsive phase behavior of the PEG–PDLA/PLLA–PEG–PLLA thermogel is irreversible and the stereocomplexed gel does not return to the solution state upon subsequent cooling.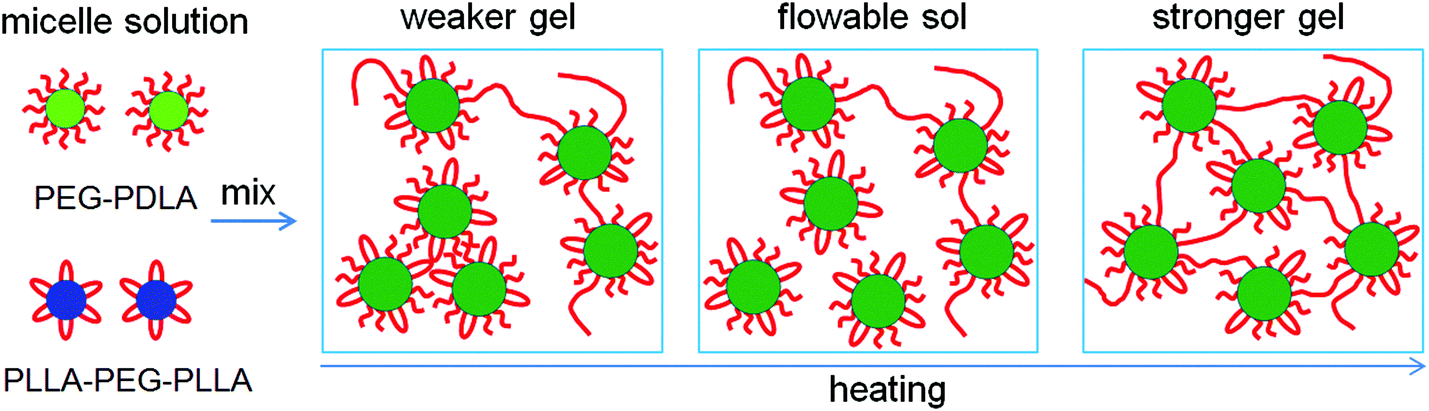 | ||
| Fig. 7 Schematic illustration for the thermally-induced gel–sol–gel transition of the PEG–PDLA/PLLA–PEG–PLLA thermogel. | ||
The rheological results of the PEG–PDLA/PLLA–PEG–PLLA mixture also support the proposed mechanism for the gel–sol–gel transition. Fig. 8 shows the modulus vs. frequency and modulus vs. strain curves of the E23D14/L14E90L14 mixture (12 wt%, 5/5) measured at 10 and 60 °C. The mixture shows frequency-dependent moduli from 0.1 to 100 Hz with a slope of ∼0.7 at 10 °C (Fig. 8A), indicating a loose crosslinking or non-crosslinking structure.23 Both G′ and G′′ of the gel decrease as the strain amplitude is over 10% at 10 °C (Fig. 8B). However, the modulus of the mixture, which was treated at 60 °C for 10 min, is almost independent of frequency in the investigated frequency region (Fig. 8C), indicating a highly-crosslinked network structure.23 For the mixture measured at 60 °C, the increase of G′′ with strain over 10% is indicative of an elastic network.47 These results demonstrate an increase in the crosslinking density of the PEG–PDLA/PLLA–PEG–PLLA mixture upon heating.
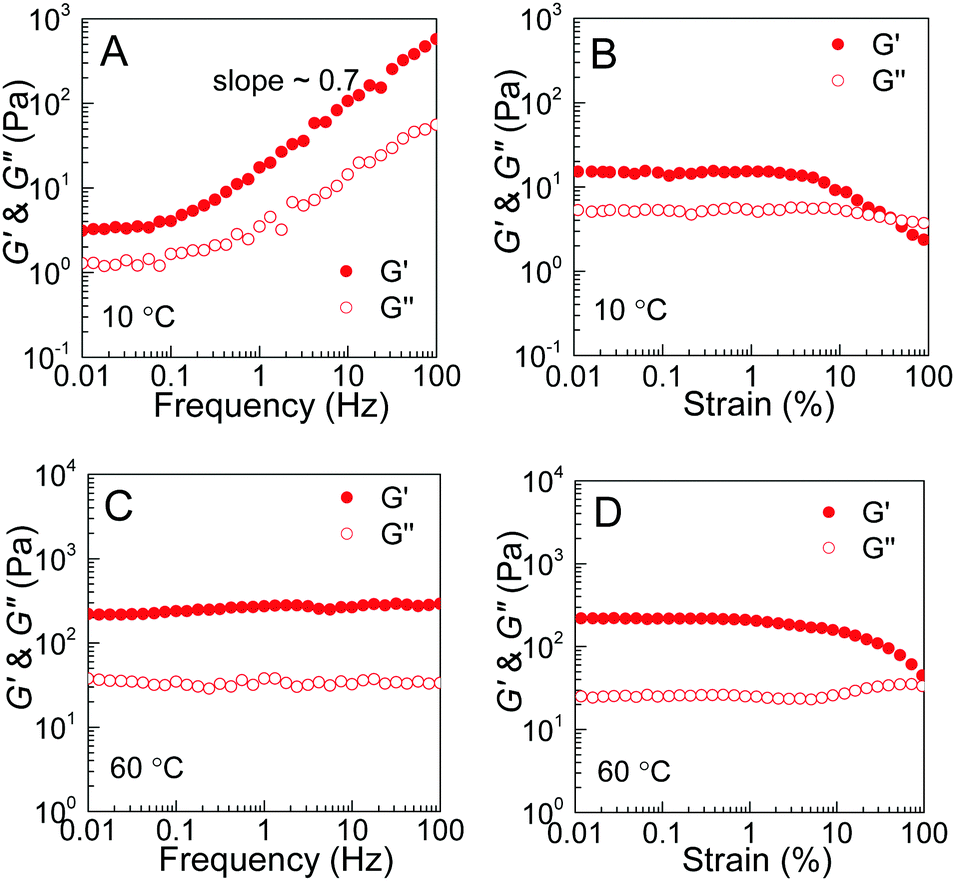 | ||
| Fig. 8 Rheological results of E23D14/L14E90L14 gels (12 wt%, 5/5): (A) G′, G′′ vs. frequency curves measured at 10 °C; (B) G′, G′′ vs. strain curves measured at 10 °C; (C) G′, G′′ vs. frequency curves measured at 60 °C; and (D) G′, G′′ vs. strain curves measured at 60 °C. PEG–PDLA and PLLA–PEG–PLLA solutions were mixed at 10 °C and equilibrated at 10 or 60 °C for 10 min before measurements. | ||
In vitro drug release behavior of stereocomplexed gels
The in vitro drug release behaviors of PEG–PDLA/PLLA–PEG–PLLA gels were investigated using DOX (a typical anticancer drug) as a model hydrophobic drug. Fig. 9 shows the DOX release profiles and representative images of E23D14/L14E45L14, E23D14/L14E90L14 and E23D14/L14E227L14 5/5 gels (15 wt%) in PB (pH 7.4, 10 mM) at 37 °C; these samples are all gels at 37 °C before the release experiment, because of the high copolymer concentration. The drug was successfully loaded into the gels during the gelation process; all the gels appeared nacarat before release (Fig. 9B). DOX was released rather slowly from the gels at the initial 48 h (Fig. 9A). The drug release from the gels proceeds mainly in two steps: (i) diffusion of the drug from the gels in the initial release stage and (ii) release of the drug with erosion of the gel matrix in the later release stage.48 Similar drug release kinetics of the gels in the initial stage of 48 h are observed, probably due to the similar drug diffusion in the gels. A sudden increase of the release rate was observed after 48 h, attributable to the drastic erosion and degradation of gels in the later stage. The release kinetics of the drug in the gels depend significantly on DPPEG,tri. As shown in Fig. 9A, the drug release rate increases in the later stage as the DPPEG,tri increases, due to the increase in the hydrophilicity of the gel and the decrease in the SC content (the SC content increases with the fraction of PLLA/PDLA blocks). The E23D14/L14E45L14 gel with small DPPEG,tri and large fSC had a slow drug release rate; only 72% of DOX was released from the gel after 96 h. The dense chain packing in stereocomplexed PLLA/PDLA domains makes the drug well encapsulated in the hydrophobic phase, thus decreasing the diffusion ability of drug molecules during the release process. On the other hand, the slower degradation rate of SCs can also prolong the release of the encapsulated drug.23 In contrast, the E23D14/L14E227L14 gel with large DPPEG,tri and small fSC% exhibited fast release and 95% of DOX was released from the gel after 96 h. As shown in Fig. 9C, the E23D14/L14E45L14 gel remained in the gel state after releasing for 96 h; the good stability of this gel was due to the higher fractions of SCs and hydrophobic domains. However, the E23D14/L14E90L14 gel became swollen and soft, whereas the E23D14/L14E227L14 gel was flowable after releasing for 96 h. Therefore, it is feasible to control the release kinetics of PEG–PDLA/PLLA–PEG–PLLA gels by varying the copolymer compositions and degree of stereocomplexation.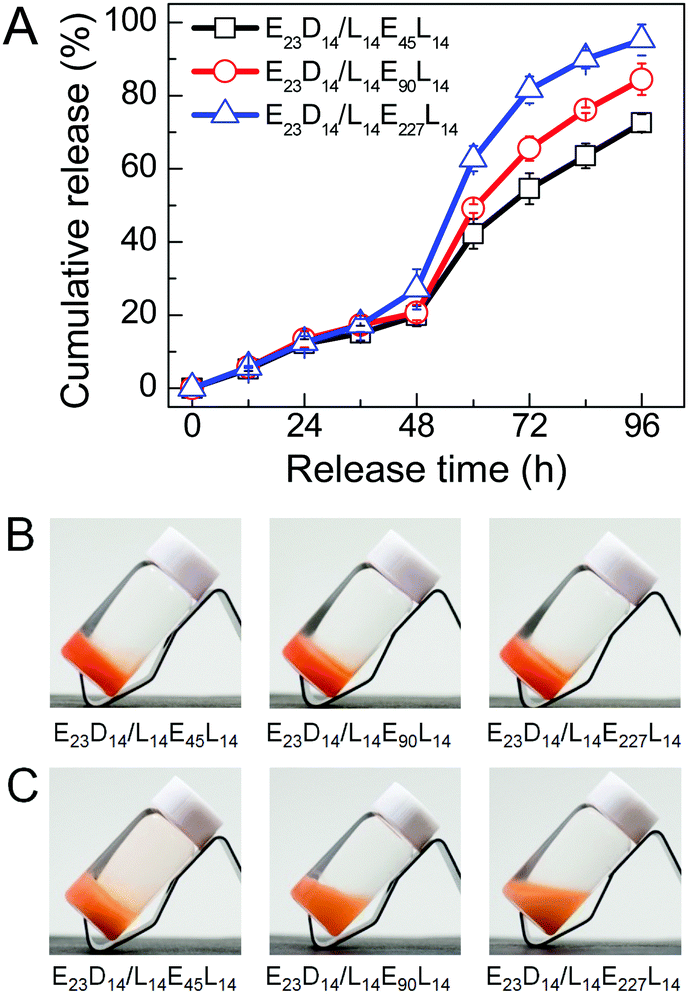 | ||
| Fig. 9 In vitro DOX-release results of PEG–PDLA/PLLA–PEG–PLLA 5/5 gels (15 wt%) in PB (pH 7.4, 10 mM): (A) drug release profiles; (B) images of DOX-loaded gels before release; and (C) images of DOX-loaded gels after releasing for 96 h. The initial content of DOX in gel was 0.04 wt%. | ||
Conclusions
In summary, PEG–PDLA/PLLA–PEG–PLLA mixtures exhibit a diversified thermoresponsive phase transition behavior, depending on the copolymer composition and mixing ratio. For the first time, we have found unique thermally-induced gel–sol–gel multiple phase transitions in the PEG–PDLA/PLLA–PEG–PLLA stereocomplexed thermogel. The microstructural changes and mechanism involved in the phase transitions of the stereocomplexed thermogel were investigated. PEG blocks show higher mobility in the solution state but are highly restricted in the gel state of the enantiomeric mixture. PLLA/PDLA stereocomplexation plays an important role in the gel–sol–gel phase transition of the thermogel; it drives the second physical gelation at higher temperature. As indicated by SAXS results, the core–shell micelle structure of the PLA/PEG block copolymer is maintained in the gel and concentrated solution, despite the strong intermicellar interactions and stereocomplexation-induced intermicellar crosslinking. It is proposed that the thermally-induced gel–sol–gel transitions originate from the synergistic effects of two aspects, i.e., (i) the increased mobility of PEG segments and micelles, and (ii) the promoted PLLA/PDLA stereocomplexation. The PEG–PDLA/PLLA–PEG–PLLA gels are able to capsulate a hydrophobic drug; the drug-loaded gels show controlled release behavior. The drug release rate can be controlled by varying the composition of the copolymers used. This study will be helpful to understand the complex phase behavior of amphiphilic copolymers in solution, and to design thermogels with the desired phase behavior and physical properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by the Natural Science Foundation of Zhejiang Province, China (LR16E030003), the Natural Science Foundation of China (21422406), and the State Key Laboratory of Chemical Engineering (SKLChE-16Z). WAXD and SAXS were performed at the BL16B1 and BL19U2 beamlines of SSRF, China, respectively. We are grateful to Dr Jan Ilavsky of the Argonne National Laboratory for the discussion on SAXS data fitting.Notes and references
- N. Rapoport, Prog. Polym. Sci., 2007, 32, 962–990 CrossRef CAS.
- M. H. Park, M. K. Joo, B. G. Choi and B. Jeong, Acc. Chem. Res., 2012, 45, 424–433 CrossRef CAS PubMed.
- L. Yu and J. Ding, Chem. Soc. Rev., 2008, 37, 1473–1481 RSC.
- B. Jeong, S. W. Kim and Y. H. Bae, Adv. Drug Delivery Rev., 2012, 64, 154–162 CrossRef.
- K. Nagahama, A. Takahashi and Y. Ohya, React. Funct. Polym., 2013, 73, 979–985 CrossRef CAS.
- A. Basu, K. R. Kunduru, S. Doppalapudi, A. J. Domb and W. Khan, Adv. Drug Delivery Rev., 2016, 107, 192–205 CrossRef CAS PubMed.
- O. Glatter, G. Scherf, K. Schillen and W. Brown, Macromolecules, 1994, 27, 6046–6054 CrossRef CAS.
- Y. K. Jung, M. H. Park, H. J. Moon, U. P. Shinde and B. Jeong, Macromolecules, 2013, 46, 4215–4222 CrossRef CAS.
- D. S. Lee, M. S. Shim, S. W. Kim, H. Lee, I. Park and T. Chang, Macromol. Rapid Commun., 2001, 22, 587–592 CrossRef CAS.
- L. Yu, H. Zhang and J. Ding, Angew. Chem., Int. Ed., 2006, 45, 2232–2235 CrossRef CAS PubMed.
- L. Chen, T. Ci, L. Yu and J. Ding, Macromolecules, 2015, 48, 3662–3671 CrossRef CAS.
- B. Jeong, Y. H. Bae and S. W. Kim, Macromolecules, 1999, 32, 7064–7069 CrossRef CAS.
- S. J. Bae, J. M. Suh, Y. S. Sohn, Y. H. Bae, S. W. Kim and B. Jeong, Macromolecules, 2005, 38, 5260–5265 CrossRef CAS.
- M. J. Hwang, J. M. Suh, Y. H. Bae, S. W. Kim and B. Jeong, Biomacromolecules, 2005, 6, 885–890 CrossRef CAS PubMed.
- W. S. Shim, J. S. Yoo, Y. H. Bae and D. S. Lee, Biomacromolecules, 2005, 6, 2930–2934 CrossRef CAS PubMed.
- A. Petit, B. Müller, R. Meijboom, P. Bruin, F. van de Manakker, M. Versluijs-Helder, L. G. J. de Leede, A. Doornbos, M. Landin, W. E. Hennink and T. Vermonden, Biomacromolecules, 2013, 14, 3172–3182 CrossRef CAS PubMed.
- J. Lee, Y. H. Bae, Y. S. Sohn and B. Jeong, Biomacromolecules, 2006, 7, 1729–1734 CrossRef CAS PubMed.
- Z. Li, Z. Zhang, K. L. Liu, X. Ni and J. Li, Biomacromolecules, 2012, 13, 3977–3989 CrossRef CAS PubMed.
- B. Jeong, Y. H. Bae, D. S. Lee and S. W. Kim, Nature, 1997, 388, 860–862 CrossRef CAS PubMed.
- B. Jeong, D. S. Lee, J.-I. Shon, Y. H. Bae and S. W. Kim, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 751–760 CrossRef CAS.
- M. S. Kim, K. S. Seo, G. Khang, S. H. Cho and H. B. Lee, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5784–5793 CrossRef CAS.
- B. Jeong, M. R. Kibbey, J. C. Birnbaum, Y.-Y. Won and A. Gutowska, Macromolecules, 2000, 33, 8317–8322 CrossRef CAS.
- H. Mao, G. Shan, Y. Bao, Z. Wu and P. Pan, Soft Matter, 2016, 12, 4628–4637 RSC.
- T. Li, T. Ci, L. Chen, L. Yu and J. Ding, Polym. Chem., 2014, 5, 979–991 RSC.
- T. Fujiwara, T. Mukose, T. Yamaoka, H. Yamane, S. Sakurai and Y. Kimura, Macromol. Biosci., 2001, 1, 204–208 CrossRef CAS.
- S. Li, Macromol. Biosci., 2003, 3, 657–661 CrossRef CAS.
- S. Li and M. Vert, Macromolecules, 2003, 36, 8008–8014 CrossRef CAS.
- T. Mukose, T. Fujiwara, J. Nakano, I. Taniguchi, M. Miyamoto, Y. Kimura, I. Teraoka and C. W. Lee, Macromol. Biosci., 2004, 4, 361–367 CrossRef CAS PubMed.
- D. G. Abebe and T. Fujiwara, Biomacromolecules, 2012, 13, 1828–1836 CrossRef CAS PubMed.
- C. W. Lee, T. Manoshiro, Y.-I. Hsu and Y. Kimura, Macromol. Chem. Phys., 2012, 213, 2174–2180 CrossRef CAS.
- K. Nagahama, K. Fujiura, S. Enami, T. Ouchi and Y. Ohya, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6317–6332 CrossRef CAS.
- C. Hiemstra, Z. Zhong, P. J. Dijkstra and J. Feijen, Macromol. Symp., 2005, 224, 119–132 CrossRef CAS.
- C. Hiemstra, Z. Zhong, L. Li, P. J. Dijkstra and J. Feijen, Biomacromolecules, 2006, 7, 2790–2795 CrossRef CAS PubMed.
- S. J. Buwalda, L. Calucci, C. Forte, P. J. Dijkstra and J. Feijen, Polymer, 2012, 53, 2809–2817 CrossRef CAS.
- H. Tsuji, Adv. Drug Delivery Rev., 2016, 107, 97–135 CrossRef CAS PubMed.
- H. Mao, P. Pan, G. Shan and Y. Bao, J. Phys. Chem. B, 2015, 119, 6471–6480 CrossRef CAS PubMed.
- C. Ma, P. Pan, G. Shan, Y. Bao, M. Fujita and M. Maeda, Langmuir, 2015, 31, 1527–1536 CrossRef CAS PubMed.
- Y.-I. Hsu, K. Masutani, T. Yamaoka and Y. Kimura, Polymer, 2015, 67, 157–166 CrossRef CAS.
- H. Tsuji and S. Yamamoto, Macromol. Mater. Eng., 2011, 296, 583–589 CrossRef CAS.
- S. K. Agrawal, N. Sanabria-DeLong, P. R. Jemian, G. N. Tew and S. R. Bhatia, Langmuir, 2007, 23, 5039–5044 CrossRef CAS PubMed.
- S. K. Agrawal, N. Sanabria-DeLong, G. N. Tew and S. R. Bhatia, Macromolecules, 2008, 41, 1774–1784 CrossRef CAS.
- D. G. Abebe, K.-Y. Liu, S. R. Mishra, A. H. F. Wu, R. N. Lamb and T. Fujiwara, RSC Adv., 2015, 5, 96019–96027 RSC.
- J. Teixeira, J. Appl. Crystallogr., 1988, 21, 781–785 CrossRef.
- P. Malo de Molina, S. Lad and M. E. Helgeson, Macromolecules, 2015, 48, 5402–5411 CrossRef CAS.
- L. Yu, Z. Zhang and J. Ding, Biomacromolecules, 2011, 12, 1290–1297 CrossRef CAS PubMed.
- C. Gong, S. Shi, L. Wu, M. Gou, Q. Yin, Q. Guo, P. Dong, F. Zhang, F. Luo, X. Zhao, Y. Wei and Z. Qian, Acta Biomater., 2009, 5, 3358–3370 CrossRef CAS PubMed.
- Y.-G. Jia and X. X. Zhu, Chem. Mater., 2015, 27, 387–393 CrossRef CAS.
- Y. Zhang, X. Wu, Y. Han, F. Mo, Y. Duan and S. Li, Int. J. Pharm., 2010, 386, 15–22 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: GPC, NMR, and DLS data and phase diagrams of PEG–PDLA, PLLA–PEG–PLLA and their mixtures. See DOI: 10.1039/c7qm00502d |
| This journal is © the Partner Organisations 2018 |