
Synthesis of porous nitrogen doped zinc oxide nanostructures using a novel paper mediated template method and their photocatalytic study for dye degradation under natural sunlight†
Gajanan
Kale
,
Sudhir
Arbuj
,
Ujjwala
Kawade
,
Sunit
Rane
,
Jalindar
Ambekar
and
Bharat
Kale
 *
*
Nanocrystalline Laboratory, Centre for Material for Electronic Technology (C-MET), Department of Information Technology, Govt. of India, Panchawati, Off Pashan Road, Pune 411007, India. E-mail: bbkale1@gmail.com; bbkale@cmet.gov.in; Fax: +91 20 2589 8180; Tel: +91 20 2589 9273
First published on 6th November 2017
Abstract
N-Doped zinc oxide (N-ZnO) nanostructures were prepared using a novel paper mediated template combustion method. The precursors were impregnated on filter papers and thermally processed to obtain porous N-ZnO nanostructures. The XRD patterns of the as-synthesized N-ZnO confirm the formation of the wurtzite phase, and the broad diffraction peaks confirm the nanocrystalline nature of the product. The TEM and SEM images show spherical shaped 20–30 nm N-ZnO nanoparticles. The UV-visible absorbance spectra of the product show a red shift in the spectra with increasing doping concentration of nitrogen in ZnO. This red shift was due to incorporation of nitrogen into the ZnO lattice. Photoluminescence spectra indicate a strong emission peak at 391 nm which corresponds to band gap emission. Considering the band gap of the as-synthesized N-ZnO to be well within the visible region, the photocatalytic activity for the degradation of methylene blue (MB) and rhodamine (RhB) dyes was studied under direct sunlight using the as-synthesized N-ZnO. The N-ZnO prepared with a higher amount of urea (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8) shows the highest photocatalytic activity as compared to the other synthesized N-ZnO samples towards the degradation of MB and RhB, i.e. complete degradation of MB in 30 min and RhB in 45 min. Our novel synthesis approach provides uniformly distributed nanosized spherical particles having higher photocatalytic activity as compared to other reported methods.
:8) shows the highest photocatalytic activity as compared to the other synthesized N-ZnO samples towards the degradation of MB and RhB, i.e. complete degradation of MB in 30 min and RhB in 45 min. Our novel synthesis approach provides uniformly distributed nanosized spherical particles having higher photocatalytic activity as compared to other reported methods.
1 Introduction
With the advent of technology, the growth rate of industries has drastically increased in all sectors such as chemical, textile, steel and power in order to fulfill the daily needs of humankind for a comfortable lifestyle. This directly or indirectly enhances the level of pollution around industrial zones.1 Industrialization and pollution are growing linearly whereas pollution abatement techniques are not seen to be so effective. The available chemical, physical and biological techniques are not cost effective and have several limitations. Also in many countries, the government has forced the stringent legislation to control pollution. For effective treatment of pollutants,2 the scientific community is in search of easy and cost effective techniques. Recently, heterogeneous semiconductor photocatalysis has emerged as a potential technique for effective abatement of pollutants3 as well as in solving energy related issues.4 In semiconductor materials having an appropriate band gap, upon illumination with light, electrons from the valence band (VB) move to the conduction band (CB) leaving holes behind. The formed electrons and holes in the CB and the VB act as stronger reducing and oxidizing agents and have the capability to reduce as well as oxidize many organic and inorganic compounds.5,6 Over the last three or four decades, the photocatalysis technique has been effectively utilized for the degradation of many pollutants present in air and water.7 TiO2 and ZnO are the best catalysts among the other studied semiconductor materials. However, the major problem with these materials is that their band gap is in the UV region and hence useful only for UV light driven photocataysis.8 For effective utilization of solar light, the band gap of these materials must be in the visible region. Many researchers have tuned the band gap of the existing semiconductors and have developed new photocatalytic materials. For enhancing the photocatalytic activity of the existing semiconductors, researchers have tried different synthesis protocols, coupled catalytic systems with metal oxides/sulphides, graphene9 and carbon nanotubes,10 noble metal loading,11 use of sacrificial reagents and so on.12 Further, cation13 and anion14 doping has been tried to tune the band gap towards the visible region. As compared to TiO2, ZnO can be easily synthesized with different morphologies such as nanorods,15 belts, nanowires16 and hierarchical nanostructures.17,18 The change in morphology19 affects the photocatalytic activity of ZnO.20 Moreover, ZnO has a wide range of applications due to its unique physical21 and chemical properties such as a wide band gap (3.37 eV), a large exciton binding energy (60 meV), thermal stability, and good piezoelectronic and optoelectronic properties.22,23 Due to these unique properties, ZnO has various applications in the areas of solar cells,24 gas sensors,25 laser diodes and thin film transistors. Considering the functionalities of ZnO, the above morphologies and nanostructures have been architectured using different methods such as sol–gel,26 hydrothermal, chemical vapour deposition,27 reactive magnetron sputtering,28 spray pyrolysis,29 pulsed laser deposition,30 high energy milling,31 sono-chemical and other wet chemical methods.9 Still the studied methods for effective nitrogen doping in ZnO in order to absorb visible light require some modifications. In this regard, we have demonstrated a simple, novel paper mediated template method for the synthesis of N-ZnO nanostructures. This method is simple and does not require any complicated equipment. Also, the ionic size of nitrogen (ionic size: 1.32 Å) and oxygen (ionic size: 1.26 Å) is comparable, leading to easy substitution of nitrogen in the place of oxygen in the ZnO crystal structure. Further, N doping significantly extends the absorption of ZnO towards the visible region. Moreover N dopants were photostable and highly soluble within the doped matrix.32Herein, we have synthesized nanostructured nitrogen doped ZnO using a novel paper mediated template-combustion technique by varying the amount of nitrogen precursor. The photocatalytic performance of the prepared N-ZnO nanostructures was investigated by following the degradation of aqueous methylene blue (MB) and rhodamine (RhB) dyes under natural sunlight.
2 Experimental section
2.1 Materials
Zinc nitrate hexahydrate (Zn(NO3)2·6H2O), urea (CO(NH2)2), methylene blue and rhodamine were obtained from Fisher Scientific International, Inc. These analytical grade precursors were used as such without any further purification. All solutions used for the synthesis were prepared with double distilled water (DW).2.2 Synthesis of N-ZnO using a paper mediated template-combustion technique
For the synthesis of ZnO, 10 mmol (2.37 gm) of zinc nitrate was dissolved in a minimum amount of distilled water. The above solution was impregnated on ashless Whatman filter paper and dried at room temperature. Further, the dried Whatman paper was heated at 150 °C for 3 h, then at 300 °C (for 3 h) and subsequently at 500 °C for 4 h. The heating rate was kept constant at 5 °C min−1 for all samples during the synthesis. The obtained ZnO was further characterized and used for the photocatalytic activity study. The same reaction procedure was followed for the synthesis of nitrogen doped ZnO (N-ZnO). A separately prepared urea solution was added to the zinc nitrate solution and stirred for 10 min to obtain a homogeneous mixture. This mixture was allowed to absorb on the Whatman filter paper, dried and heated at 150, 300, and 500 °C as mentioned above. The same synthesis approach was repeated by varying the amount of urea in order to dope a higher amount of nitrogen in ZnO. The ZnO synthesized using zinc nitrate by changing the molar concentration of urea as 1:0, 1:4, 1:8, and 1:10 was labeled G0, G4, G8 and G10, respectively.
2.3 Characterization
The UV-visible absorbance spectra of the ZnO and N-ZnO nanostructures were measured with a Shimadzu UV-3600 spectrophotometer in the spectral range of 200–800 nm using the diffuse reflectance mode. Photoluminescence (PL) spectroscopy was performed with a Shimadzu (RF-5301 PC) spectrofluorometer. The crystal structures of the ZnO and N-ZnO nanostructures were examined by the powder X-ray diffraction technique (XRD, Bruker Advanced D8) using a Cu Kα radiation source. The crystalline size was calculated usingScherrer's formula d = 0.9λ/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) cosθ cosθ |
2.4 Photocatalytic activity study
The photocatalytic activity of the prepared ZnO and N-ZnO nanostructures was studied using degradation of aqueous MB and RhB dyes (10 ppm) under solar light. For this purpose, 100 mg of the powder was dispersed in a 250 mL conical flask containing 100 mL of 10 ppm dye solution and stirred under solar light. Before keeping the suspension under solar light, this suspension was magnetically stirred in the dark for 60 min to establish an adsorption–desorption equilibrium between the photocatalyst and the dye. Once the suspension was kept under solar light, a small amount of the sample was removed at fixed intervals of time and centrifuged, and the supernatant liquid was analysed with a UV-visible spectrophotometer. The absorbance at 664 and 555 nm was used to calculate the amount of MB and RhB dyes degraded with time respectively. The Degradation % is calculated using the following formula:| Degradation % = [(C0 − Ct)/C0] × 100 |
C0/Ctvs. irradiation time.
3 Results and discussion
3.1 Structural study
The X-ray diffraction patterns of the synthesized ZnO and N-ZnO nanostructures with varying urea concentrations are depicted in Fig. 1. Pure ZnO shows the formation of a highly crystalline hexagonal phase (Fig. 1, G0). N doped ZnO prepared with varying urea concentrations also shows the formation of a hexagonal structure. The diffraction peaks clearly match with those of the standard ICDD pattern (no. 36-1451) which confirms the formation of the wurtzite phase of ZnO. The broader XRD peaks also indicate the nanocrystalline nature of N-ZnO. The correlation between the urea concentration and the crystal structure of N-ZnO is not observed. The crystallite size measured by Scherrer's equation was found to be 21, 17, 19 and 21 nm, for the G0, G4, G8 and G10 samples, respectively. | ||
| Fig. 1 XRD patterns of pure ZnO (G0) and N doped ZnO (G4, G8 and G10). | ||
3.2 Optical study
The UV-visible diffuse reflectance spectra (UV-DRS) of ZnO (G0) and N-ZnO (G4, G8, G10) are shown in Fig. 2(a). Pristine ZnO depicts an absorption edge around 388 nm which corresponds to a band gap of 3.19 eV. N-ZnO shows enhanced absorbance in the range of 400 to 700 nm. The absorbance of the G8 sample is more towards the visible light region than the other prepared samples i.e. G8 > G4 > G10 > G0. This observed red shift in the absorbance indicates the possibility of nitrogen doping in the ZnO structure. The G8 sample prepared using a mole ratio of 1:8 (zinc nitrate:urea) has higher absorbance in the visible region as it may provide a sufficiently higher amount of nitrogen for doping. Whereas, further use of a higher urea concentration (G10) leads to a higher reaction temperature during the combustion process, affecting the removal of doped nitrogen from the ZnO structure. This might be the reason for loading a lower amount of nitrogen even after using a higher amount of urea for the synthesis of N-ZnO.36
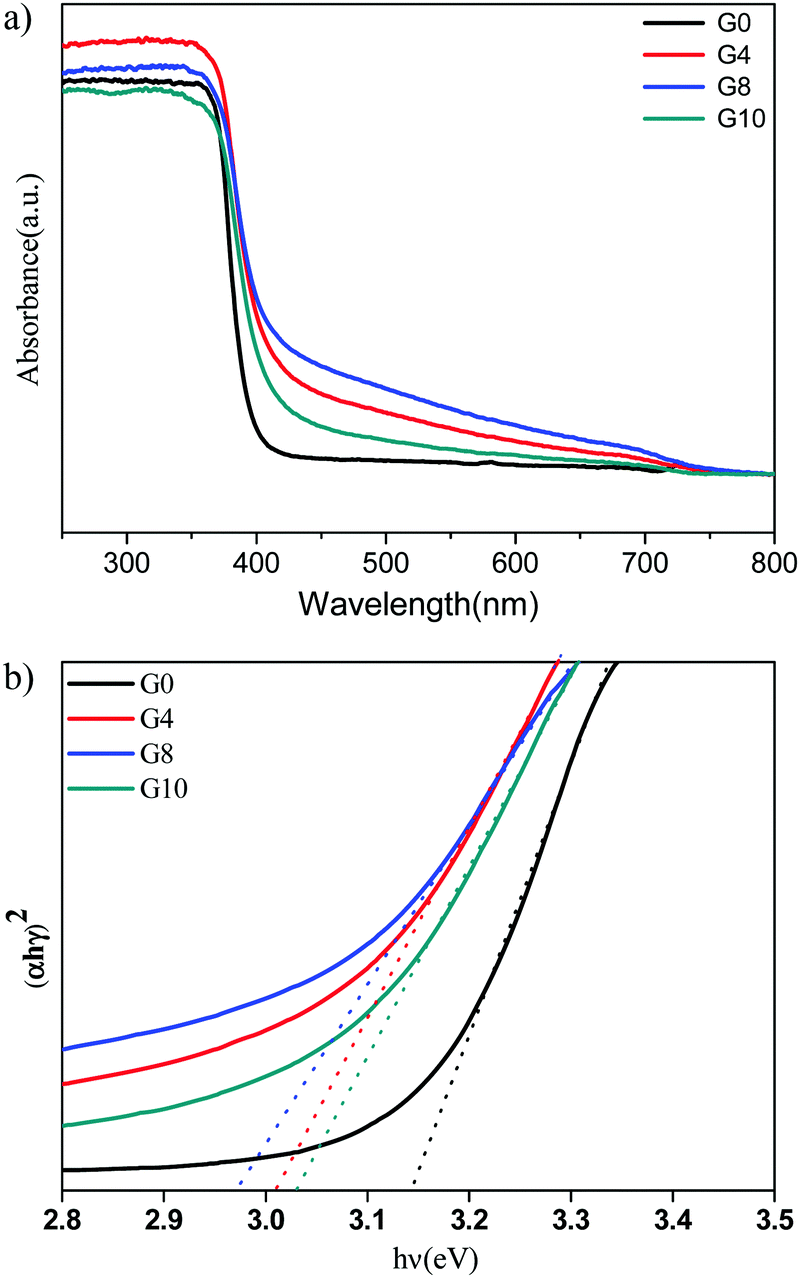 | ||
| Fig. 2 (a) UV-DRS spectrum of pure and nitrogen doped ZnO G0 (pure), G4 (1:4) G8 (1:8) and G10 (1:10). (b) Tauc's plots for ZnO (G0 – 3.14 eV) and N-ZnO (G4 – 3.01, G8 – 2.97, G10 – 3.03 eV). | ||
The Tauc plots for the estimation of band gap values of N-ZnO are shown in Fig. 2(b).
The observed band gap values for G0, G4, G8 and G10 are 3.14, 3.01, 2.97 and 3.03 eV respectively. Overall, the red shift in the absorbance depicts nitrogen doping in the ZnO structure.
The room temperature PL spectra of ZnO and N-ZnO nanostructures were taken at an excitation wavelength of 350 nm in an aqueous medium (Fig. 3). For PL measurements, 1 mg of the sample was dispersed in 10 mL of distilled water and then employed for measurement. The PL spectra of pure ZnO show a strong emission peak at 391 nm which corresponds to band edge emission and a slight hump at 470 nm attributed to surface defects. In the case of N-ZnO, the intensity of the band edge emission peak at 391 nm decreased with increase in the amount of urea. This may be due to the creation of mid gap vacancies by nitrogen doping.37 This also inhibits charge carrier recombination.
 | ||
| Fig. 3 Photoluminescence (PL) spectra of ZnO pure-G0 and N-doped G4, G8 and G10. | ||
3.3 XPS study
X-ray photoelectron spectroscopy (XPS) analysis of the G8 sample was carried out in order to confirm the chemical composition of the N-ZnO nanostructure (Fig. 4).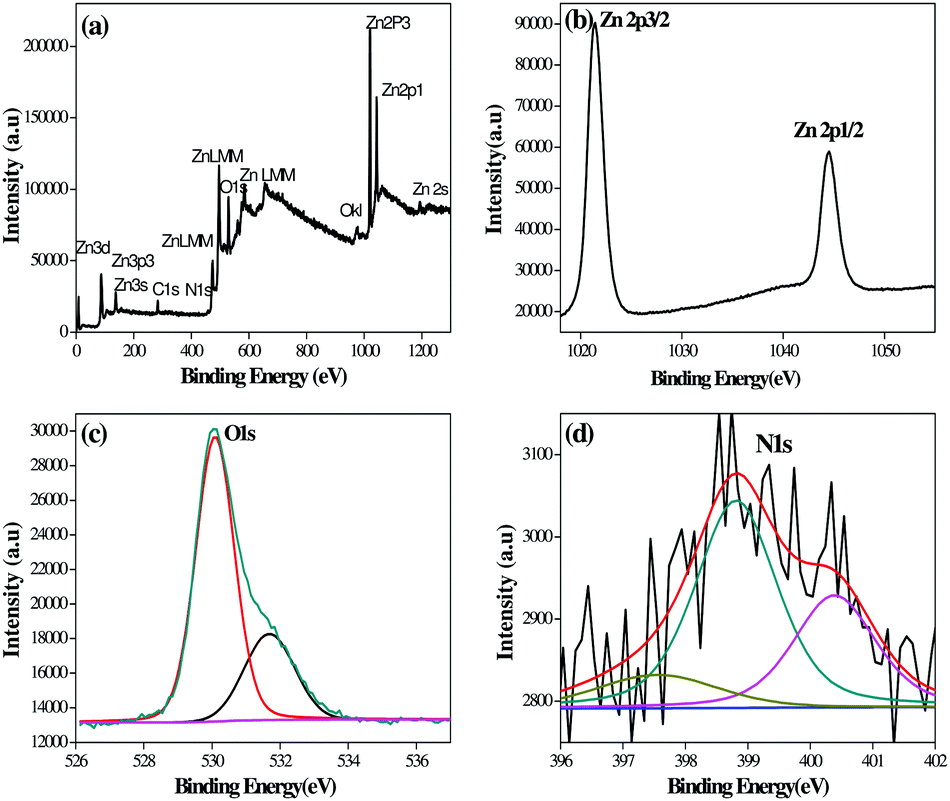 | ||
| Fig. 4 XPS spectra of the N-ZnO (G8) sample, (a) full range XPS spectra containing Zn, C, O and N, (b) resolution spectra for Zn, (c) resolution spectra for O and (d) resolution spectra for N. | ||
The full range XPS spectra containing Zn, C, O and N (Fig. 4(a)) along with high-resolution spectra for Zn, O and N are depicted in Fig. 4(b)–(d) respectively. The carbon element present in the spectra is due to adventitious hydrocarbons from the XPS instrument.38 The bands at 1021.38 and 1044 eV correspond to Zn 2p3/2 and Zn 2p1/2 respectively.39 The high resolution spectra after O 1s deconvolution show that oxygen exists in two forms i.e. 530.09 and 531.68 eV. The bands at 530.09 eV correspond to oxygen in ZnO.40 The bands at 531.68 eV indicate other oxygen containing species, such as O2 and OH− group on the ZnO surface.41,42Fig. 4(d) shows N1 bands at 397.55, 398.81 and 400.39 eV which indicate the presence of N-ZnO. The peak at 397.55 eV corresponds to substitutional doping of nitrogen in place of oxygen in the ZnO lattice. The peak at 398.81 eV corresponds to interstitial doping in the crystal lattice associated with anionic N− in metallic nitrites (N-Zn binding).43 The peak above 400 eV is generally attributed to NO− or NO2− type of species. In our case, the peak observed at 400.39 eV may correspond to these species.44 The XPS study clearly shows the presence of nitrogen in the ZnO lattice.
3.4 Morphological study
The surface and morphological characteristics play a vital role in governing the catalytic performance of the material. The prepared ZnO and N-ZnO nanostructures were analyzed using FE-SEM and the micrographs are depicted in Fig. 5. | ||
| Fig. 5 FE-SEM images of samples (a and b) G0-pure ZnO, (c and d) G4 (1:4) N-ZnO, (e and f) G8 (1:8) N-ZnO and (g and h) G10 (1:10) N-ZnO. | ||
Pure ZnO indicates the formation of spherical particles having a uniform distribution with a particle size in the range of 20–30 nm. The particles are well connected with each other forming a dense network (Fig. 5a and b) along with a porous nature. The G4 sample having a urea concentration of 1:4 indicates the formation of spherical morphology with enhanced linkage among the spherical particles (Fig. 5c and d). The porous layered sheets are created by the alignment of the same sized (20–30 nm) nanoparticles. Further, increase in the urea concentration in the case of the G8 sample results in the retention of spherical morphology of the formed N-ZnO with a denser network among the N-ZnO particles. Furthermore, in the G10 sample with a urea concentration of 1:10, the nanosized N-ZnO particles agglomerated to form a sub-micron sized chunk like morphology. The particles are well connected to each other forming continuous sheet like structures. Overall, with increase in the urea concentration, there is no difference in the particle size of ZnO but formation of a denser network is observed.
The low and high resolution FETEM images of the prepared pure ZnO and N-ZnO nanostructures are shown in Fig. S3 (ESI†) and Fig. 6 respectively, along with the selected area electron diffraction pattern. Pure ZnO depicts the formation of spherical shaped particles along with oval and distorted spherical type morphologies (S3). For the G8 sample uniform particle size distributions in the range of 20–30 nm were observed (Fig. 6).
 | ||
| Fig. 6 FETEM images of nitrogen doped ZnO (G8), ZnO/urea molar ratio: 1:8, with the SAED pattern. | ||
TEM images of N-ZnO also depict the same kind of morphologies as observed in the case of G4 samples (ESI,† Fig. S4). Further, increase in the concentration of urea confirms the formation of distorted spherical type morphologies with an increase in the particle size. The cluster of small particles creates a submicron sized network. The FETEM depicts an inter-planar spacing of 0.248 nm which corresponds to the (101) hkl plane of hexagonal ZnO. The bright spots in the SAED pattern validate the formation of highly crystalline hexagonal N-ZnO nanostructures and corroborate the XRD results.
3.5 Photocatalytic activity
The photocatalytic study was performed using MB and RhB. We have chosen MB and RhB dyes for degradation as they are one of the major constituents in textile wastes. For the photocatalytic performance study of the prepared ZnO and N-ZnO nanostructures, we have selected an ideal composition, i.e. 100 mg of the catalyst and 100 mL of 10 ppm aqueous dye solution. With reference to our earlier results, we observed that with an increase in the catalyst amount, the dye degradation rate increased up to a certain level and after an optimum concentration the rate decreased. This decrease in the rate of photoactivity was due to the increase in opacity of the solution resulting in penetration of a lower number of photons in our existing photoreactor system. One more reason may be the aggregation of solid particles, resulting in scattering of light.35The photocatalytic study was carried out in sunny days during the months of April–May between 11 am and 3 pm under natural sunlight. The graph of % methylene blue vs. irradiation time is depicted in Fig. 7(a).
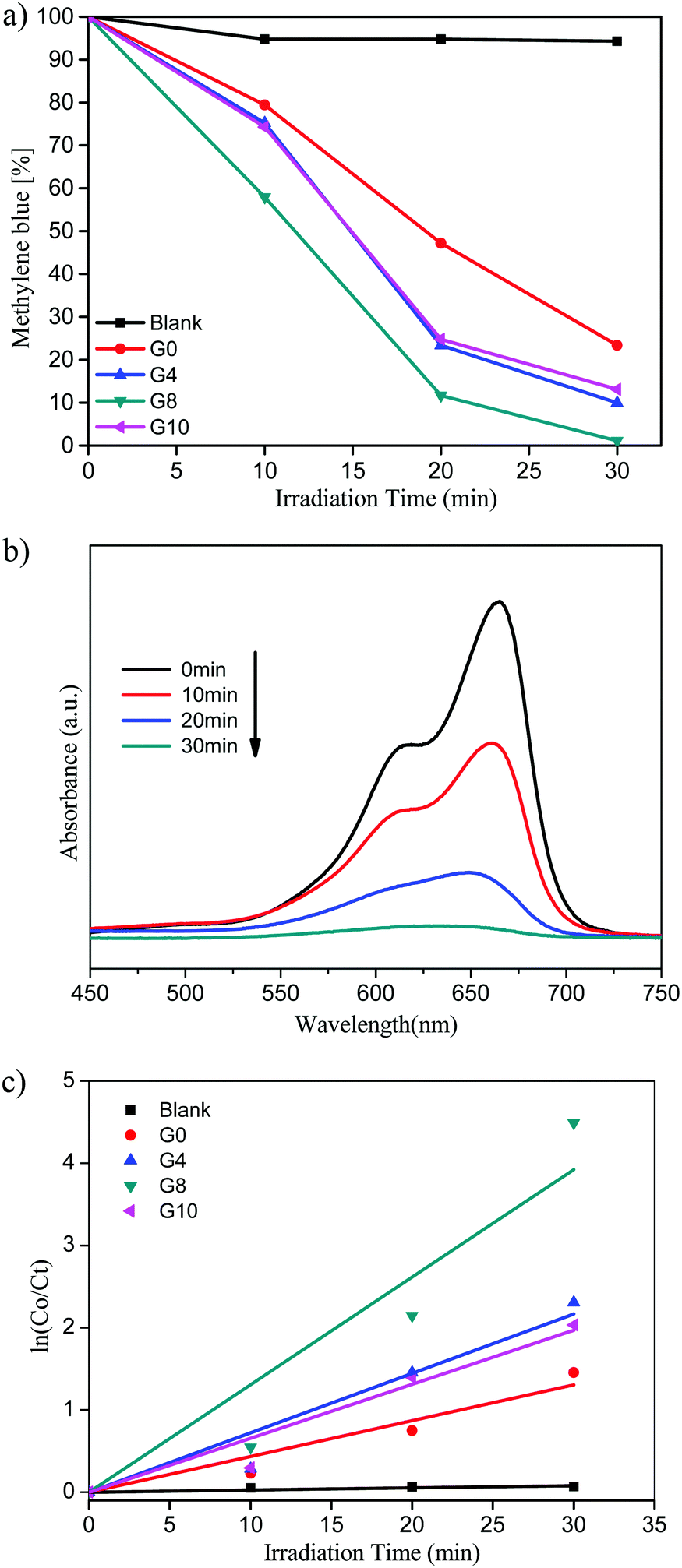 | ||
| Fig. 7 (a) Methylene blue (%) vs. irradiation time. (b) UV-visible absorbance of MB for the G8 sample. (c) Kinetic study for MB degradation. | ||
Pure ZnO shows 77% of MB degradation within 30 min, whereas the G8 sample completely degraded the MB solution under the same experimental conditions. The other catalysts prepared with varying urea concentrations G4 and G10 show 90 and 87% of MB degradation within 30 min, respectively.
The UV-visible absorbance of MB with time using the G8 sample is depicted in Fig. 7(b). From these spectra, the % MB recovered was calculated with respect to irradiation time. The G8 sample shows the highest photocatalytic activity among the prepared compositions due to higher doping of N in the ZnO lattice. N-ZnO (G8) has extended absorbance in the visible region (400 to 700 nm) resulting in absorption of more number of photons, which in turn generates more electron–hole pairs. This enhanced absorbance leads to the generation of more electron–hole pairs resulting in higher photocatalytic activity towards MB degradation. The photocatalytic activity trend of ZnO and N-ZnO is G8 > G4 > G10 > G0. In the case of the G10 sample, even though a higher amount of urea was used, due to lower doping of nitrogen it exhibited poor photocatalytic performance compared to the other prepared compositions. Generally, the photocatalytic dye degradation reaction follows pseudo-first order reaction kinetics. The chemical kinetics of dye degradation reactions are studied using the Langmuir–Hinshelwood model. The slope of the graph of lnC0/Ctvs. irradiation time gives the value of apparent rate constant (kapp).45 The graph of lnC0/Ctvs. irradiation time is depicted in Fig. 7(c). The photocatalytic MB degradation rates using ZnO and N-ZnO catalysts are tabulated in Table 1. The G8 sample shows an apparent rate constant value (kapp) of 13.8 × 10−2 min−1 for MB dye degradation.
| Sr. no. | Catalyst | MB | RhB | ||
|---|---|---|---|---|---|
| Rate constant (kapp) for MB degradation (min−1) | Standard error | Rate constant (kapp) for RhB degradation (min−1) | Standard error | ||
| 1 | Blank | 0.27 × 10−2 | ±0.04 × 10−2 | 0.18 × 10−2 | ±0.03 × 10−2 |
| 2 | G0 | 4.3 × 10−2 | ±0.43 × 10−2 | 2.15 × 10−2 | ±0.18 × 10−2 |
| 3 | G4 | 7.2 × 10−2 | ±0.7 × 10−2 | 4.04 × 10−2 | ±0.13 × 10−2 |
| 4 | G8 | 13.8 × 10−2 | ±1.6 × 10−2 | 7.79 × 10−2 | ±1.15 × 10−2 |
| 5 | G10 | 6.5 × 10−2 | ±0.5 × 10−2 | 3.27 × 10−2 | ±0.28 × 10−2 |
The same trend was observed for RhB dye degradation using the prepared ZnO and N-ZnO nanostructures. Fig. 8(a) depicts the graph of % rhodamine vs. irradiation time.
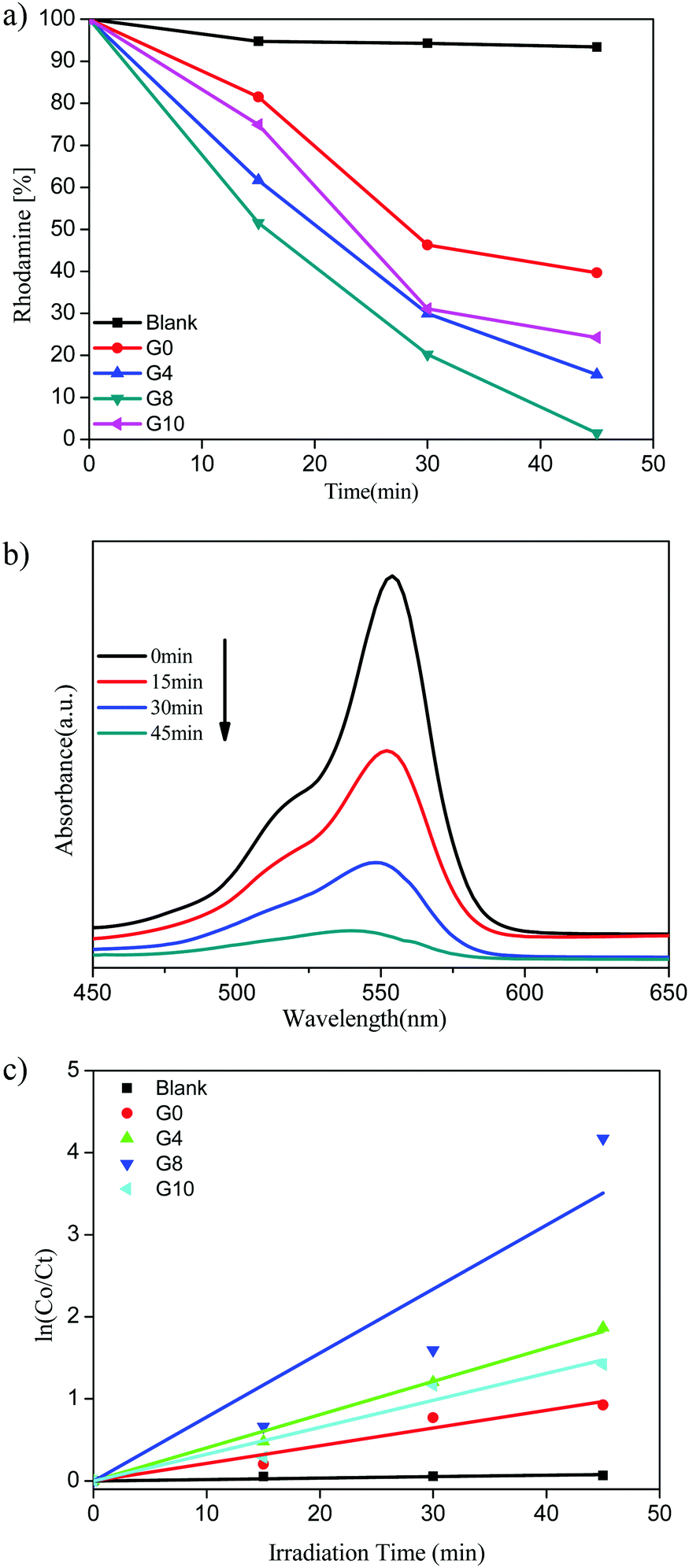 | ||
| Fig. 8 (a) Rhodamine (%) vs. irradiation time. (b) UV-visible absorbance of RhB for the G8 sample. (c) Kinetic study for RhB degradation. | ||
The highest photocatalytic activity for RhB dye degradation was observed for sample G8 (99% degradation) in 45 min. At the same time the degradation using ZnO and N-ZnO was found to be 61%, 85% and 76% for samples G0, G4 and G10, respectively.
The UV-visible absorbance spectra of the RhB dye with time using the G8 sample is depicted in Fig. 8(b). The rate of reaction for RhB degradation was calculated from the slope of the graph of lnC0/Ctvs. irradiation time shown in Fig. 8(c). Also the rate constant values for RhB dye degradation are depicted in Table 1. As per the rate constant value (kapp), the G8 sample shows the highest rate constant value of 7.79 × 10−2 min−1 for the RhB dye. As compared to MB degradation, the RhB dye required a quite higher reaction time for degradation. This might be due to the complex structure of the RhB dye than that of MB.
Overall, sample G8 shows higher activity due to higher doping of nitrogen in ZnO as explained above. A photoluminescence study shows a decrease in the intensity of emission peaks, due to vacancies created by doping. A higher number of charge carriers are generated due to more absorption of visible light, and at the same time there is a suppression of charge carrier recombination due to vacancies created by nitrogen doping.9 Hence the higher photocatalytic activity obtained for sample G8 is quite justifiable. As compared to other combustion methods32 N-ZnO prepared using this method shows higher photocatalytic activity and particles are uniformly distributed with size in the range of 20–30 nm. N-ZnO synthesized using this approach exhibits a porous nanostructure which may also be the reason for its good activity.
A reusability study was also performed which shows similar degradation activity. The recovered 90 mg of catalyst G8 (from the 1st run) was reused for the degradation of MB and RhB.
Degradation was completed within 33 and 48 min for MB and RhB, respectively (ESI,† Fig. S1 and S2). This reusability study shows the same activity under similar conditions which shows the good stability of the catalyst. In a nutshell, the new synthesis approach is quite useful in obtaining highly crystalline nitrogen doped semiconductors and functionality as a photocatalyst.
4 Conclusion
We have successfully prepared uniformly distributed ZnO and N-ZnO nanostructures using a paper-mediated template technique for the first time. ZnO shows enhanced absorbance in the visible region with an increase in the urea concentration up to 8 mole%. Above a certain level (10%), the absorption edge again shifted to a lower wavelength. Our method provides a new and easier technique to dope nitrogen in the ZnO lattice that can effectively utilize solar radiation. Due to nitrogen doping there is creation of vacancies which suppress charge carrier recombination and ultimately enhance the photocatalytic activity. The reusability study clearly shows the stable photocatalytic activity. The present synthesis approach will have the potential to produce other nano-structured oxides.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Ministry of Electronics and Information Technology (MeitY), Government of India, for financial support and C-MET Pune for providing research facilities. They would like to thank the Nanocrystalline Materials Group for kind support.Notes and references
- B. Crathorne, Y. J. Ress, S. France and R. M. Harrison, Pollution: Causes, Effects and Control (4), The Royal Society of Chemistry, 2001, pp. 1–31 Search PubMed.
- P. M. Birgani, N. Ranjbar, R. C. Abdullah, K. T. Wong, G. Lee, S. Ibrahim, C. Park, Y. Yoon and M. Jang, J. Environ. Manage., 2016, 184, 229–239 CrossRef CAS PubMed.
- F. Chen, Q. Yang, X. Li, G. Zeng, D. Wang, C. Niu, J. Zhao, H. An, T. Xie and Y. Deng, Appl. Catal., B, 2016, 200, 330–342 CrossRef.
- S. Aditya Kiran, Y. LukkaThuyavan, G. Arthanareeswaran, T. Matsuura and A. F. Ismail, Chem. Eng. J., 2015, 286, 528–537 CrossRef.
- J. C. Cardoso, G. G. Bessegato, J. F. de Brito, B. r. C. A. Souza and M. V. B. Zanoni, Recent Advances in Complex Functional Materials: From Design to Application, Springer International Publishing, Cham, 2017, pp. 239–269 Search PubMed.
- Z. Yang, X. Xu, X. Liang, C. Lei, Y. Cui, W. Wu, Y. Yang, Z. Zhang and Z. Lei, Appl. Catal., B, 2017, 205, 42–54 CrossRef CAS.
- B. Srikanth, R. Goutham, R. Badri Narayan, A. Ramprasath, K. P. Gopinath and A. R. Sankaranarayanan, J. Environ. Manage., 2017, 200, 60–78 CrossRef CAS PubMed.
- P. N. Paulino, V. M. M. Salim and N. S. Resende, Appl. Catal., B, 2015, 185, 362–370 CrossRef.
- A. P. Bhirud, S. D. Sathaye, R. P. Waichal, L. K. Nikam and B. B. Kale, Green Chem., 2012, 14, 2790–2798 RSC.
- V. Georgakilas, D. Gournis, V. Tzitzios, L. Pasquato, D. M. Guldi and M. Prato, J. Mater. Chem., 2007, 17, 2679–2694 RSC.
- S. T. Kochuveedu, Y. H. Jang and D. H. Kim, Chem. Soc. Rev., 2013, 42, 8467–8493 RSC.
- F. Opoku, K. K. Govender, C. G. C. E. van Sittert and P. P. Govender, New J. Chem., 2017, 41, 11701–11713 RSC.
- Ş. Ş. Türkyilmaz, N. Güy and M. Özacar, J. Photochem. Photobiol., A, 2017, 341, 39–50 CrossRef.
- J. Zhang, X. Zhang, S. Dong, X. Zhou and S. Dong, J. Photochem. Photobiol., A, 2016, 325, 104–110 CrossRef CAS.
- G. Meenakshi, A. Sivasamy, G. A. Suganya Josephine and S. Kavithaa, J. Mol. Catal. A: Chem., 2016, 411, 167–178 CrossRef.
- C. Soci, A. Zhang, B. Xiang, S. A. Dayeh, D. P. R. Aplin, J. Park, X. Y. Bao, Y. H. Lo and D. Wang, Nano Lett., 2007, 7, 1003–1009 CrossRef CAS PubMed.
- L. Xu, Q. Chen and D. Xu, J. Phys. Chem. C, 2007, 111, 11560–11565 CAS.
- J. Y. Lao, J. Y. Huang, D. Z. Wang and Z. F. Ren, J. Mater. Chem., 2004, 14, 770–773 RSC.
- A. Hezam, K. Namratha, Q. A. Drmosh, B. N. Chandrashekar, K. K. Sadasivuni, Z. H. Yamani, C. Cheng and K. Byrappa, CrystEngComm, 2017, 19, 3299–3312 RSC.
- F. Vines, O. Lamiel-Garcia, F. Illas and S. T. Bromley, Nanoscale, 2017, 9, 10067–10074 RSC.
- D. Mudusu, K. R. Nandanapalli, S. R. Dugasani, S. H. Park and C. W. Tu, Sci. Rep., 2016, 6, 28561 CrossRef PubMed.
- S. C. Dixon, S. Sathasivam, B. A. D. Williamson, D. O. Scanlon, C. J. Carmalt and I. P. Parkin, J. Mater. Chem. C, 2017, 5, 7585–7597 RSC.
- M. M. Rahman, M. K. R. Khan, M. R. Islam, M. A. Halim, M. Shahjahan, M. A. Hakim, D. K. Saha and J. U. Khan, J. Mater. Sci. Technol., 2012, 28, 329–335 CAS.
- J. Falgenhauer, F. Fiehler, C. Richter, M. Rudolph and D. Schlettwein, Phys. Chem. Chem. Phys., 2017, 19, 16159–16168 RSC.
- A. Paliwal, A. Sharma, M. Tomar and V. Gupta, Sens. Actuators, B, 2017, 250, 679–685 CrossRef CAS.
- H. Qin, W. Li, Y. Xia and T. He, ACS Appl. Mater. Interfaces, 2011, 3, 3152–3156 CAS.
- C. L. Perkins, S.-H. Lee, X. Li, S. E. Asher and T. J. Coutts, J. Appl. Phys., 2005, 97, 034907 CrossRef.
- Y. Nakano, T. Morikawa, T. Ohwaki and Y. Taga, Appl. Phys. Lett., 2005, 87, 232104 CrossRef.
- J.-L. Zhao, X.-M. Li, J.-M. Bian, W.-D. Yu and C.-Y. Zhang, J. Cryst. Growth, 2005, 280, 495–501 CrossRef CAS.
- J. R. Duclère, M. Novotny, A. Meaney, R. O'Haire, E. McGlynn, M. O. Henry and J. P. Mosnier, Superlattices Microstruct., 2005, 38, 397–405 CrossRef.
- J. Lu, Q. Zhang, J. Wang, F. Saito and M. Uchida, Powder Technol., 2006, 162, 33–37 CrossRef CAS.
- H. Sudrajat and S. Babel, Journal of Water Process Engineering, 2017, 16, 309–318 CrossRef.
- S. S. Arbuj, R. R. Hawaldar, U. P. Mulik, B. N. Wani, D. P. Amalnerkar and S. B. Waghmode, J. Mater. Sci. Eng. B, 2010, 168, 90–94 CrossRef CAS.
- M. S. SolmazAghdasi, Iran. J. Catal., 2016, 6(5), 481–487 Search PubMed.
- P. Sangpour, F. Hashemi and A. Z. Moshfegh, J. Phys. Chem. C, 2010, 114, 13955–13961 CAS.
- A. K. Kulkarni, C. S. Praveen, Y. A. Sethi, R. P. Panmand, S. S. Arbuj, S. D. Naik, B. B. Kale and A. V. Ghule, Dalton Trans., 2017, 46, 14859–14868 RSC.
- A. Bhirud, S. Sathaye, R. Waichal, C.-J. Park and B. Kale, J. Mater. Chem. A, 2015, 3, 17050–17063 CAS.
- D. Zhang, J. Gong, J. Ma, G. Han and Z. Tong, Dalton Trans., 2013, 42, 16556–16561 RSC.
- I. M. P. Silva, G. Byzynski, C. Ribeiro and E. Longo, J. Mol. Catal. A: Chem., 2016, 417, 89–100 CrossRef CAS.
- S. Anandan, A. Vinu, K. L. P. Sheeja Lovely, N. Gokulakrishnan, P. Srinivasu, T. Mori, V. Murugesan, V. Sivamurugan and K. Ariga, J. Mol. Catal. A: Chem., 2007, 266, 149–157 CrossRef CAS.
- X. H. Wang, S. Liu, P. Chang and Y. Tang, Phys. Lett. A, 2008, 372, 2900–2903 CrossRef CAS.
- C. Shifu, Z. Sujuan, L. Wei and Z. Wei, J. Hazard. Mater., 2008, 155, 320–326 CrossRef PubMed.
- H. Ma, X. Cheng, C. Ma, X. Dong, X. Zhang, M. Xue, X. Zhang and Y. Fu, Int. J. Photoenergy, 2013, 8 Search PubMed.
- M. Sathish, B. Viswanathan, R. P. Viswanath and C. S. Gopinath, Chem. Mater., 2005, 17, 6349–6353 CrossRef CAS.
- S. S. Arbuj, U. P. Mulik and D. P. Amalnerkar, Nanosci. Nanotechnol. Lett., 2013, 5, 968–973 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qm00490g |
| This journal is © the Partner Organisations 2018 |