
Modulation of luminescence chromic behaviors and environment-responsive intensity changes by substituents in bis-o-carborane-substituted conjugated molecules†
Hiroki
Mori
,
Kenta
Nishino
,
Keisuke
Wada
,
Yasuhiro
Morisaki‡
 ,
Kazuo
Tanaka
* and
Yoshiki
Chujo
*
,
Kazuo
Tanaka
* and
Yoshiki
Chujo
*
Department of Polymer Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Nishikyo-ku, Kyoto 615-8510, Japan. E-mail: tanaka@poly.synchem.kyoto-u.ac.jp; chujo@poly.synchem.kyoto-u.ac.jp
First published on 29th January 2018
Abstract
Two types of multi-functional emissive bis-o-carborane-substituted 1,4-bis(phenylethynyl)benzene molecules were synthesized, and their optical properties were investigated in detail. The pristine o-carborane-substituted molecule CBH simultaneously exhibited dual emission from the locally excited (LE) and twisted intramolecular charge transfer (TICT) states in solution. Originating from changes in the intensity ratios between both emission bands, clear solvatochromic and thermochromic behaviors were observed. Surprisingly, TICT emission was observed even in the solid state. Aggregation- and crystallization-induced emission enhancement (AIEE and CIEE, respectively) were also presented by CBH. These solid-state emission enhancements could be derived from the suppression of aggregation-caused quenching (ACQ) by the bulky cage structure and the spherical shape of o-carborane. Next, we also synthesized the methyl-substituted derivative (CBMe) and found environment-resistant highly-efficient emission in both the solution and solid states. Finally, CBMe presented mechanochromic luminescence in the solid state. The substituent effects on the optical properties are discussed.
Introduction
Incorporation of optically-functional “element-blocks”, which are defined as a minimum functional unit containing heteroatoms,1,2 is a promising strategy not only for obtaining bright luminescent materials but also for showing stimuli-responsive characteristics. For example, a series of luminescent conjugated molecules and polymers has recently been developed based on luminescent boron “element-blocks”.3–5 By introducing aggregation-induced emission (AIE)-active “element-blocks”6–8 into the conjugated main-chain, solid-state luminescence9–13 and AIE properties14,15 were realized in the “element-block polymers”. Furthermore, by employing stimuli-responsive luminescent “element-blocks”, chemical sensors and environment-sensitive luminescent chromism were also accomplished.16–23 Therefore, discovery and exploration of unique characteristics from “element-blocks” is a topic with high relevance, especially in material science as well as in fundamental photochemistry.From this stand point, o-carborane,24–29 which is a cluster compound containing two carbon and ten boron atoms, is a potential “element-block”30–32 for constructing a conjugated system because of its unique solid-state luminescence properties.33–51 Moreover, it is known that the o-carborane units work as a strong electron acceptor when connected at the carbon.52,53 Therefore, by combination with electron-donating aryl units, intense emission from the intramolecular charge transfer (ICT) state was often obtained.52 In particular, it has been revealed that o-carborane can play a significant role in presenting solid-state emission in various systems by suppressing ACQ.33–51 It was proposed that the steric structure of o-carborane should play a critical role in avoiding ACQ by disturbing intermolecular interactions with the chromophore unit. Owing to this advantage of the o-carborane unit, this ICT emission can often be detected from o-carborane derivatives with high emission efficiencies even in the solid state where emission properties were often spoiled via the ACQ process.54–58 Furthermore, it was suggested from theoretical investigation that the electronic state of the o-carborane unit should vary by its rotation.59 Especially, the electron-accepting ability of o-carborane critically depends on the angle between the C–C bond in the o-carborane unit and the aryl substituent.52 Based on this fact, luminescent chromism has been accomplished with π-conjugated aryl-substituted o-carborane structures.60,61 Thus, the construction and evaluation of new conjugation systems connected with o-carborane are of great significance to discover unique photochemical phenomena as well as to develop advanced optical materials.
Herein, we present the syntheses and optical properties of bis-o-carborane-substituted 1,4-bis(phenylethynyl)benzene having multi-functional emissive properties. Two types of o-carborane derivatives with or without the methyl substituent at the adjacent carbon to the aryl moiety were prepared for evaluating the influence of molecular motion on electronic structures. From optical measurements in the solution and solid states, it was found that drastic changes can be induced by the substituents. In the absence of the substituents, dual emission from the LE and TICT states in the solution state, AIEE, CIEE and solvato- and thermochromism were observed. In contrast, the o-carborane derivative having methyl substituents showed constant intense emission in both the solution and solid states and mechanochromic luminescence behavior. These various useful luminescence properties can be explained by the degree of molecular motion and the structures being controlled by the substituent effect.
Results and discussion
According to Scheme 1, bis-o-carborane and methylated o-carborane-substituted 1,4-bis(phenylethynyl)benzene CBH and CBMe were synthesized, respectively. It was presumed that the molecular rotation at the o-carborane units would be disturbed by the methyl substituent at the adjacent position. The two steps of the Sonogashira–Hagihara coupling reactions from 1,4-dibromo-2,5-diiodobenzene (1) to 1,4-bis(2′-phenylethyn-1′-yl)-2,5-bis(2′-trimethylsilylethyn-1′′-yl)benzene (3a) followed by the deprotection of K2CO3 afforded 1,4-diethynyl-2,5-bis(2′-phenylethyn-1′-yl)benzene (3a′). Then, treatment with decaborane (B10H14) in the presence of N,N-dimethylaniline and the successive regioselective alkyne-insertion reaction afforded CBH. Similarly, CBMe was also synthesized by the two steps of the Pd-catalyzed coupling reactions followed by the alkyne-insertion reaction with decaborane, although the reaction yield was low due to steric hindrance. CBH and CBMe were stable to H2O, air, and heat in both the solution and solid states at least for half a year. The structures of the obtained compounds were characterized by 1H, 11B and 13C NMR spectroscopies and high-resolution mass analyses (Charts S1–S6, ESI†).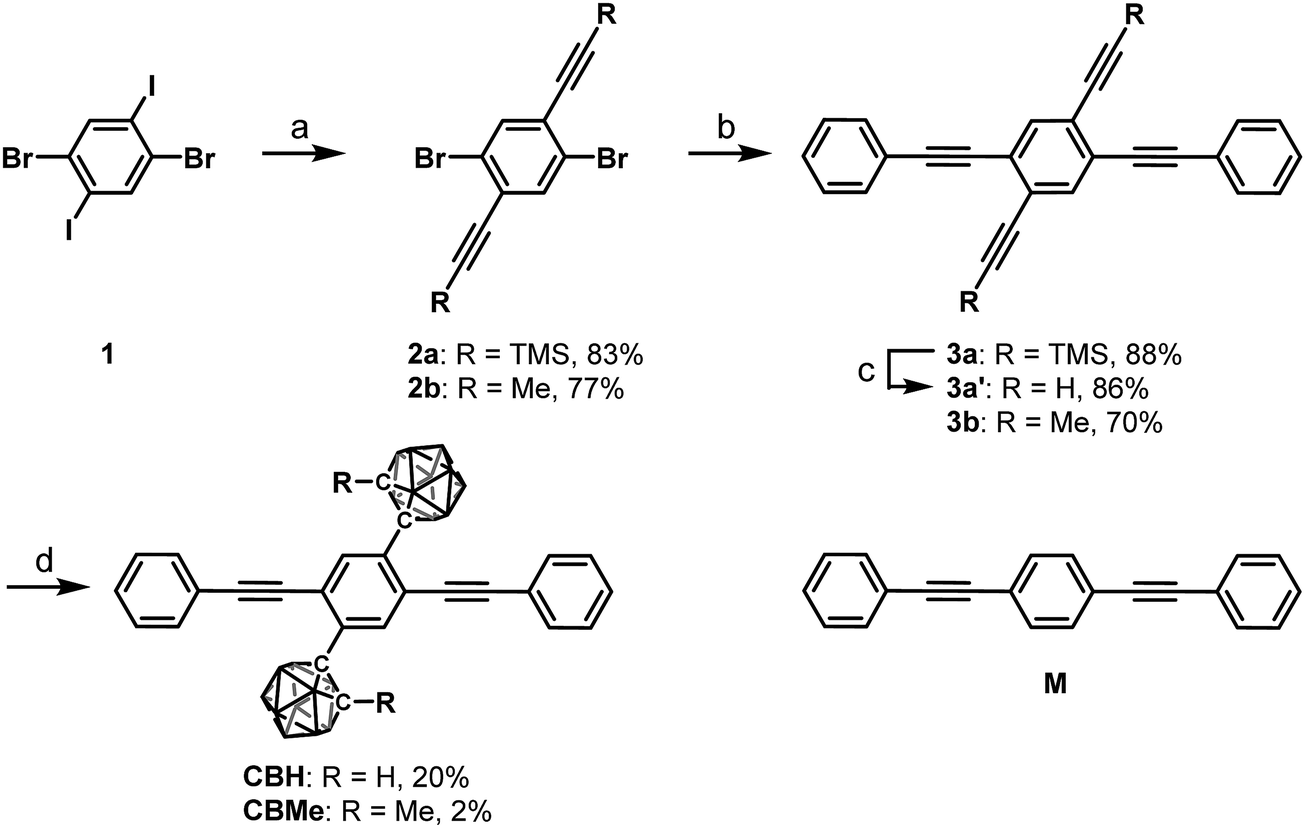 | ||
Scheme 1 Synthesis of CBH and CBMe. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reagents and conditions: (a) ethynyl compound, Pd(PPh3)4, CuI, THF, iPr2NH, r.t., 36 h; (b) phenylacetylene, Pd(PPh3)4, CuI, THF, Et3N, reflux, 22 h for 3a, 48 h for 3b; (c) K2CO3, THF, MeOH, r.t., 4 h; (d) decaborane, N,N-dimethylaniline, toluene, reflux, 7 d for CBH, 5 d for CBMe. Reagents and conditions: (a) ethynyl compound, Pd(PPh3)4, CuI, THF, iPr2NH, r.t., 36 h; (b) phenylacetylene, Pd(PPh3)4, CuI, THF, Et3N, reflux, 22 h for 3a, 48 h for 3b; (c) K2CO3, THF, MeOH, r.t., 4 h; (d) decaborane, N,N-dimethylaniline, toluene, reflux, 7 d for CBH, 5 d for CBMe. | ||
The structure of CBH was successfully confirmed by the X-ray crystallographic analysis (Fig. 1, Table S1, ESI†). The dihedral angle C2–C1–C3–C4 (φ) of CBH was 21°. This fact indicates that the 1,4-bis(phenylethynyl)benzene moiety has approximately a co-planar structure including the C1–C2 bond. In the crystal packing diagrams, CBH showed π-stacks with a distance of 4.190 Å between the phenyl rings of 1,4-bis(phenylethynyl)benzene moieties. It was presumed that steric repulsion of bulky icosahedral carborane clusters could be responsible for these structures.
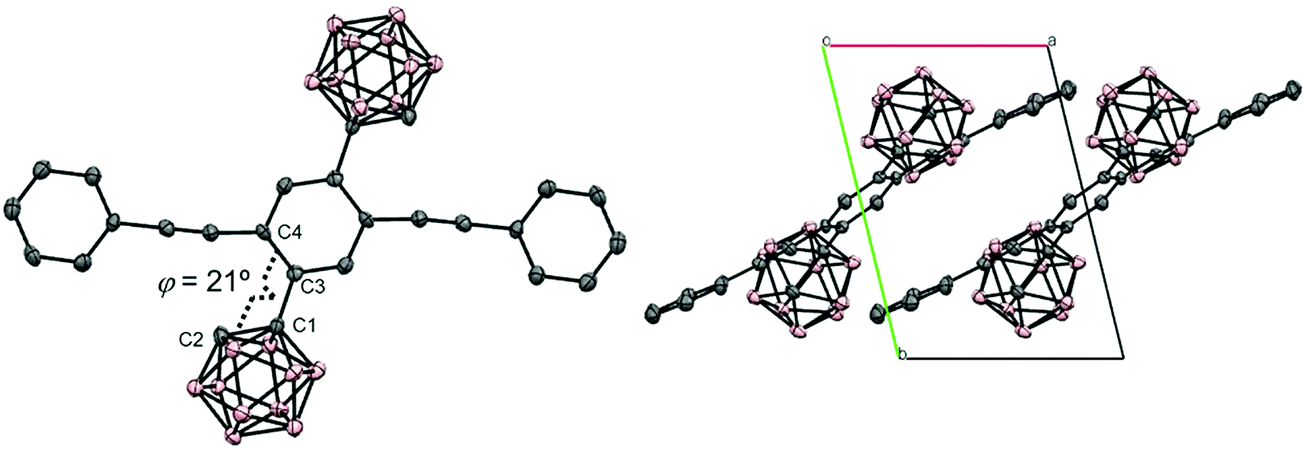 | ||
| Fig. 1 Molecular structure and packing diagrams of CBH (hydrogen atoms are omitted for clarity, and thermal ellipsoids are displayed at 30% probability). | ||
The electronic properties of CBH in the ground state were examined by UV-vis absorption measurements. Fig. 2a shows the UV-vis absorption spectra of CBH and the model compound M (1,4-bis(phenylethynyl)benzene) in THF solution (1.0 × 10−5 M). CBH exhibited a large absorption band in the UV region (ε > 35000 M−1 cm−1) assigned to the π–π* transition in the 1,4-bis(phenylethynyl)benzene moiety that corresponded to that of M. As listed in Table 1, the absorption band of CBH (λmax = 344 nm) was obtained in a longer wavelength region compared to that of M (λmax = 320 nm). It was proposed that the effect of the strong electron-accepting character of o-carborane can contribute to constructing a significant electronic interaction between the conjugated moiety and the o-carborane unit, followed by the peak shift via extension of π-conjugation to the whole molecule.
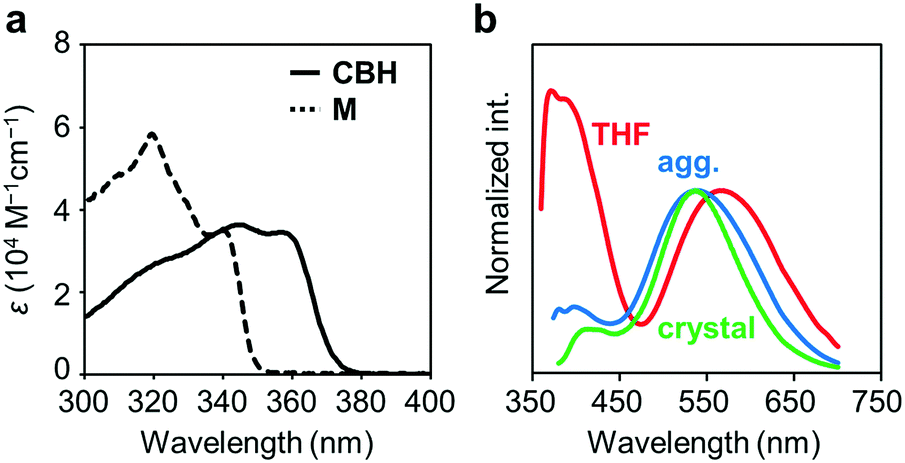 | ||
| Fig. 2 (a) UV-vis absorption spectra of CBH and M (1.0 × 10−5 M) and (b) normalized PL spectra of CBH in THF solution (1.0 × 10−5 M), aggregates (THF/H2O v/v = 1/99 solution, 1.0 × 10−5 M) and the crystal. | ||
| THFa | Aggregationb | Crystal | |||||||
|---|---|---|---|---|---|---|---|---|---|
| λ abs [nm] | ε [M−1 cm−1] | E g [eV] | λ em [nm] | Φ PL | λ em [nm] | Φ PL | λ em [nm] | Φ PL | |
| a Measured in THF solution (1.0 × 10−5 M). b Measured in THF/H2O v/v = 1/99 solution (1.0 × 10−5 M). c Calculated from the onset value in the absorption spectra. d Determined as an absolute value with the integration sphere method. e Not detectable. | |||||||||
| CBH | 344 | 36300 |
3.33 | 371, 566 | 0.30 | 538 | 0.36 | 537 | 0.57 |
| CBMe | 368 | 39200 |
3.23 | 540 | 0.84 | 542 | 0.81 | 493 | 0.78 |
| M | 320 | 58500 |
3.55 | 347 | 0.95< | —e | —e | —e | —e |
Next, the emission properties of CBH were investigated. The most impressive point was the dual-emissive property of CBH in the solution (Fig. 2b). Sharp emission bands with vibrational structures and broad ones were observed with peaks at around 370 nm and 570 nm, respectively. According to the previous reports, it was presumed that the aryl-modified o-carboranes can present ICT emission.52 To elucidate the emission mechanism of CBH, optical spectra in various solvents (toluene, CHCl3, EtOAc, THF, CH2Cl2, DMF and MeCN) were measured, and the Lippert–Mataga plots were prepared (Fig. 3a and Fig. S1, Table S2, ESI†). By increasing solvent polarity, solvatochromism was detected only from the bands around 570 nm. These data including the degree of Stokes shifts calculated with the position of the absorption bands clearly indicate that the emission bands around 370 nm and 570 nm are from the LE and ICT states, respectively. Lower emission efficiencies were obtained from the modified carboranes than from M. It is known that molecular motions including rotation at the connecting bond to the aryl moiety and vibration at the C–C bond in the o-carborane unit in the solution state should induce excitation deactivation along non-radiation processes.30 Thus, emission annihilation was observed.
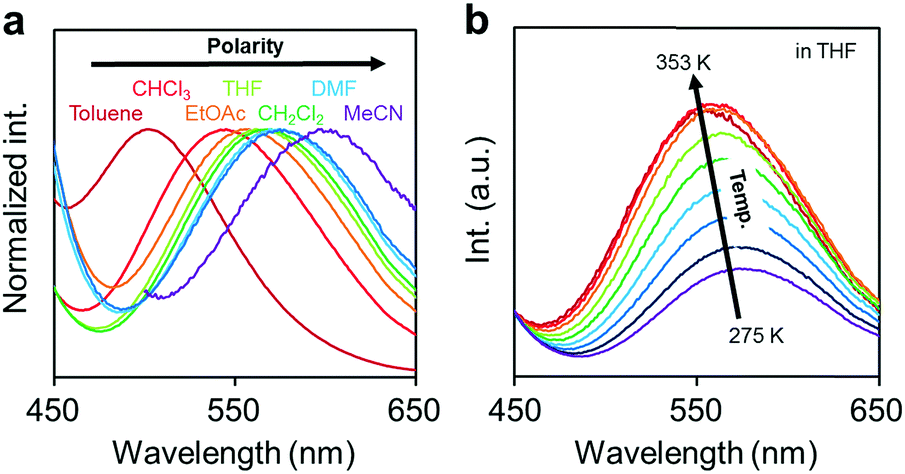 | ||
| Fig. 3 (a) Solvent and (b) temperature-dependent peak shifts in the PL spectra of the solutions containing CBH (1.0 × 10−6 M). | ||
To evaluate the TICT character, the influence of molecular rotation on luminescence properties was examined by changing temperature. Thermochromic luminescence properties were observed. From the spectra in Fig. 3b, it was shown that the intensity of the emission band from the ICT state was enhanced by heating. This is reasonable because the transition from LE to ICT should be assisted by heating. Furthermore, it should be noted that only the LE emission was detected in the frozen state at 77 K where the molecular structure should be fixed at the initial state (Fig. S2, ESI†). This result means that structural alteration from the planar conformation, which can provide the LE emission, should proceed in the excited state followed by the exhibition of the ICT emission. From these data, the luminescence mechanism of CBH can be summarized. In the ground state, the planar conformation is dominant, leading to the LE emission. On exposure to photo-excitation, molecular rotation occurs, resulting in the formation of the ICT state. Finally, the emission band with environmental responsivity in the longer wavelength region was observed. Due to steric hindrance of the 1,4-bis(phenylethynyl)benzene moiety, rotation was disturbed to some extent. Thus, the dual emissive property was obtained.
The solid-state emission properties of CBH were evaluated (Fig. 2b and Table 1). Interestingly, larger emission efficiencies were observed in both the aggregation state prepared by adding water to the THF solution and in the crystalline state. These data confirm that CBH has AIEE and CIEE properties. Similarly to the previous reports on aryl-modified o-carboranes, the o-carborane units should inhibit intermolecular motions in the condensed state because of steric hindrances. Thereby, ACQ was suppressed. In the previous report, it was shown that the formation of the TICT state was allowed even in the crystalline state. To evaluate the possibility of solid-state TICT formation with CBH, a photoluminescence (PL) spectrum was monitored at 77 K (Fig. S3, ESI†). Obviously, the emission band from the ICT state disappeared and only the LE emission was obtained. The same discussion based on the TICT mechanism should be applicable to that in the solution.
Next, by introducing the methyl groups at the adjacent carbon in the o-carborane units, the influence on optical properties was investigated. Synthesis was performed according to Scheme 1, and the characterization data indicated that the product has the desired structure. Unfortunately, a suitable single-crystal sample for X-ray crystallography was not obtained. However, it was suggested that the molecular structure should be fixed to the twisted conformation, which is similar to the structure in the TICT state.
Several comparison studies were carried out for evaluating the influence of the substituent effect on electronic properties. Fig. 4a shows the UV-vis absorption spectra of CBMe in THF solution (1.0 × 10−5 M). The sharp peak around 260 nm and the broad peak around 370 nm assigned to the π–π* transition band of the 1,4-bis(phenylethynyl)benzene moieties were observed. The values of the optical band gaps (Eg), which were estimated from the onset wavelength of the UV-vis absorption spectra, were in the order of CBH > CBMe (Table 1). The LUMO energy level was estimated from cyclic voltammetry (Fig. S4, ESI†) peak onset potentials, and the HOMO energy level was calculated from the LUMO energy level and the band gap energy estimated from the absorption edge (Table S3, ESI†). CBH and CBMe showed low-lying LUMO energy levels, −3.06 eV and −3.18 eV, respectively. By introducing the adjacent methyl groups into the o-carborane units, the molecular structure is anchored at the twisted conformation. According to the previous work, the electron-accepting ability of o-carborane is maximized at the twisted conformation.52 Thus, a low-lying LUMO was realized, as observed in the cyclic voltammograms, leading to the narrower band-gap energy of CBMe than that of CBH.
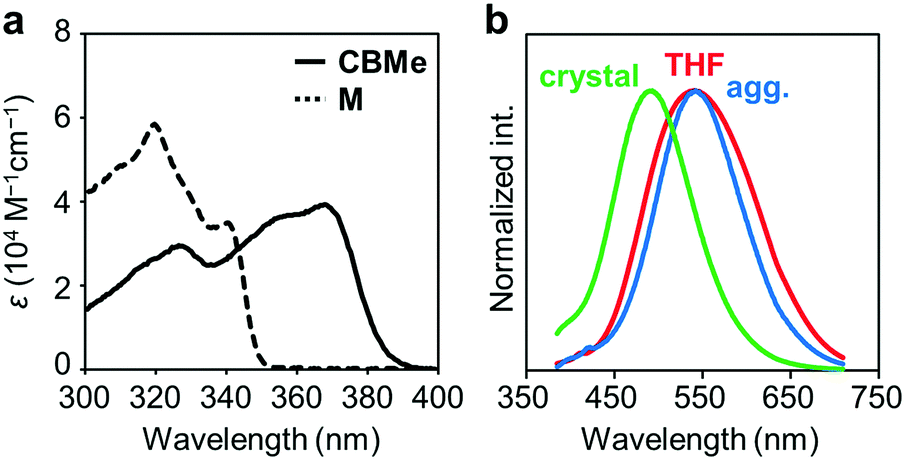 | ||
| Fig. 4 (a) UV-vis absorption spectra of CBMe and M (1.0 × 10−5 M) and (b) normalized PL spectra of CBMe in THF solution (1.0 × 10−5 M), aggregates (THF/H2O v/v = 1/99 solution, 1.0 × 10−5 M) and the crystal. | ||
The emission properties of CBMe were evaluated in various states (Fig. 4b). From the THF solution, a single emission band was observed with a peak at around 550 nm. From the PL spectra in various solvents followed by the analysis with the Lippert–Mataga plots, it was indicated that this broad emission band should result from the ICT state (Fig. S5, ESI†). Interestingly, CBMe showed high quantum efficiencies in the aggregate state (ΦPL = 0.81), the crystalline state (ΦPL = 0.78) and even in solution (ΦPL = 0.84). It is assumed that excitation deactivation induced by the intramolecular motion at the o-carborane unit could be restricted by the substituent effect.55 Furthermore, the molecular structure was fixed at the twisted conformation. As a consequence, ICT should be efficiently induced in the excited state. This speculation can be supported by the result from the solid-state emission at 77 K (Fig. S6, ESI†). Significant changes were hardly obtained by cooling the crystalline sample of CBMe. Molecular motions should be effectively suppressed by the methyl substituent.
Another notable point was the difference in emission wavelengths between the amorphous and crystalline samples (Fig. 4b and Table 1). It should be mentioned that the crystalline sample of CBMe provided the emission band in a shorter wavelength region than those in the amorphous state as well as in solution. In other words, CBMe was expected to present environment-sensitive luminescence properties. By taking into consideration this assumption, the mechanochromic luminescence properties were examined with CBMe. The crystalline sample was pounded in a mortar until the detectable peaks were eliminated in the X-ray diffraction analysis (Fig. S7, ESI†). During the mechanical treatments, the optical spectra were monitored (Fig. 5). Initially, the emission band was observed at 493 nm (ΦPL = 0.78). It was shown that the peak position was shifted to a longer wavelength region by the treatment (λem = 516 nm, ΦPL = 0.61). These data clearly indicate that CBMe has mechanochromic luminescence properties. Compared with the aggregation experiment with CBMe, as shown in Fig. 4, the emission band shift proceeded to the reverse direction by the formation of an amorphous state. In the solution-based system, solvent polarity should be responsible for emission properties. Therefore, the blue-shift might be induced by being surrounded by hydrophobic molecules. In the solid state, the degree of molecular packing should play a critical role in the electronic states. Because of bulky units including o-carboranes and methyl groups in CBMe, intermolecular interactions might be disturbed in the crystal packing, and blue-shifted emission was obtained. By the mechanical treatment, the molecular distribution should be randomized, and subsequently stabilization of energy levels by π-stacking followed by red-shifted emission could be realized. This scenario was also supported by the blue-shifted excitation spectrum. In the ground state, non-specific interactions could be formed. In the commodity mechanochromic luminescent materials, critical reduction of emission efficiencies was often induced after grinding, whereas CBMe presented a slight reduction of emission efficiency. The spherical molecular shape of o-carborane should be responsible for exhibiting environment-resistant intense emission by suppressing ACQ.
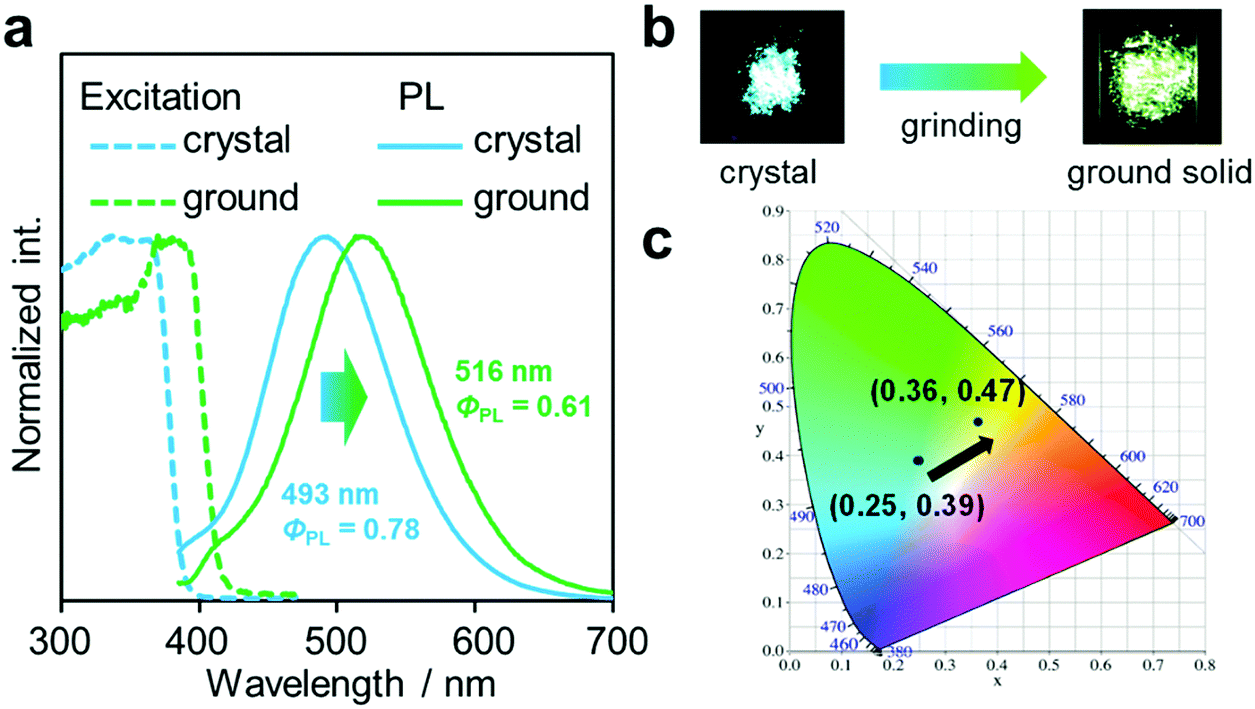 | ||
| Fig. 5 (a) PL and excitation spectra of the solid samples of CBMe. (b) Pictures of the solid samples under UV irradiation before and after grinding. (c) Luminescent colors of the samples on the CIE diagram. | ||
To deeply understand the TICT behavior of CBH, quantum chemical calculations were performed at the B3LYP/6-31+G(d)//B3LYP/6-31+G(d) level. Two geometries having planar (φ = 180°) and twisted (φ = 96°) conformers were obtained from the estimation for the optimized S1 structure of CBH. The molecular orbitals involved in the electronic transition are shown in Fig. 6. It was found that the S1–S0 electronic transition in both conformers should be mainly derived from the lowest unoccupied molecular orbital (LUMO) to the highest occupied molecular orbital (HOMO); 95% for the planar and 98% for the twisted conformers. HOMOs in both conformers and the LUMO in the planar conformer were mainly on the 1,4-bis(phenylethynyl)benzene moiety, while the LUMO in the twisted conformer was significantly delocalized to the o-carborane unit. These results strongly support that the emission from the LE and ICT states was derived from the planar and twisted conformers, respectively. The calculated emission wavelengths for the LE and TICT states were 400 nm and 733 nm, respectively. In addition, the C–C bond length in the o-carborane unit was 1.66 Å in the planar conformer and 2.40 Å in the twisted conformer, indicating that stronger electron-withdrawing should occur in the twisted conformer.52 This fact corresponded to the ICT character of the emission band from the twisted conformer. Finally, we evaluated the possibility of solid-state TICT in the crystalline state. From the calculation with the structure determined by X-ray crystallography (φ = 21°), it was shown that the HOMO and LUMO were localized mainly at the 1,4-bis(phenylethynyl)benzene moiety. Moreover, according to the experimental result, the calculated emission wavelength was 395 nm. From these data, it was proposed that CBH can exhibit only emission from the LE state. Indeed, the corresponding emission band was obtained at 77 K; meanwhile, the emission from the ICT state was mainly observed in the far longer wavelength region (Fig. S3, ESI†). These data strongly suggest that CBH should form the TICT excited state even in the crystalline state.
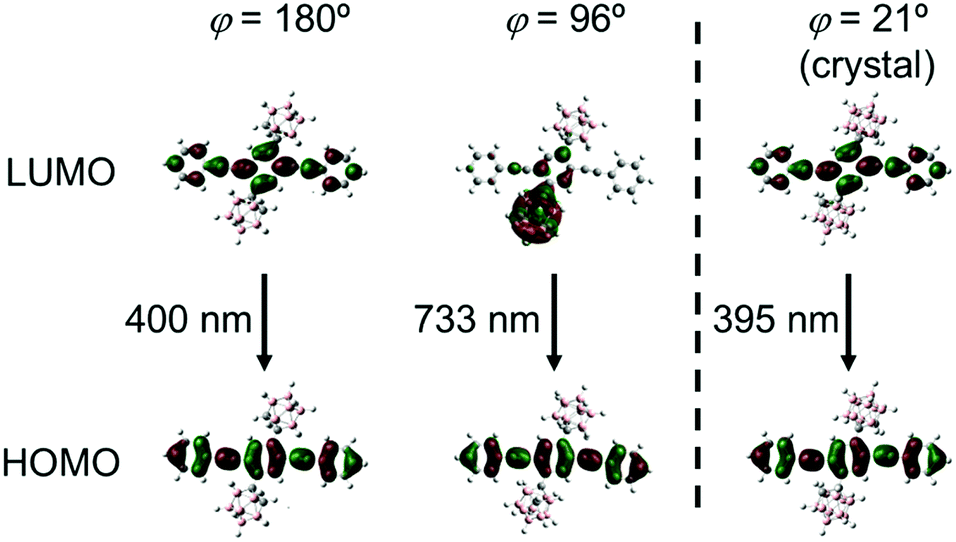 | ||
| Fig. 6 Frontier orbitals involved in the S1–S0 transition for CBH at different 1,4-bis(phenylethynyl)benzene orientations (φ = 180°, 96° and 21°). | ||
The S0–S1 electronic transition was investigated by using the same level of calculation (Fig. 7). Compared with the model compound M, the energy levels of both the HOMO and LUMO of CBH were lower by 0.56 eV and 0.76 eV, respectively. The large decreases in energy levels of the frontier orbitals could be presumably derived from the strong electron-withdrawing character of the o-carborane unit, resulting in a lower band gap than M. These results showed good agreement with the red-shift of the absorption maximum and electrochemical data of CBH (Fig. 2 and Fig. S4, ESI†).
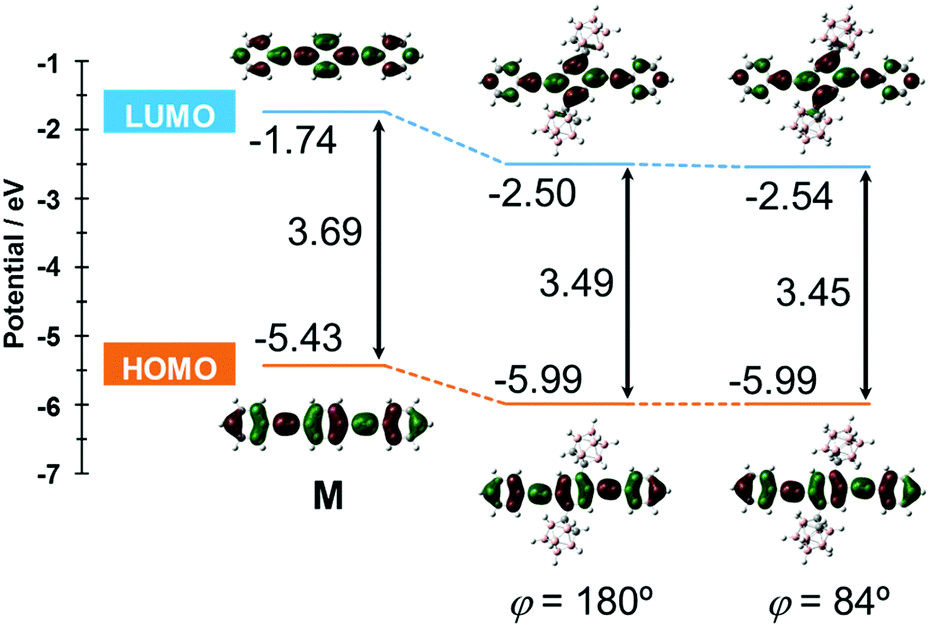 | ||
| Fig. 7 Frontier orbitals involved in the S0–S1 transition and their energy levels for CBH and M at the B3LYP/6-31+G(d) level of theory. | ||
To support the optical properties of CBMe, DFT and TD-DFT calculations were carried out at the B3LYP/6-31+G(d)//B3LYP/6-31+G(d) level (Fig. S8 and S9, ESI†). From the structural optimization, an almost perpendicular conformation to the C–C bond in o-carborane (φ = 97°) was obtained and the transition wavelength was almost the same as that in the absorption spectrum. As shown in the results from the TD-DFT calculation, the frontier orbitals involved in the S1–S0 transition were mainly derived from the LUMO to HOMO transition (98%). The HOMO was found mostly on the 1,4-bis(phenylethynyl)benzene unit, while the LUMO was localized on the o-carborane unit with substantial orbital contribution, which is similar to the case of CBH (Fig. 6). These results strongly suggest that the emission around 490 nm originates from the ICT excited state. In addition, the LUMO partly existed on the C–H bond in the methyl group. This fact implies that the electron donation of the C–H antibonding orbital (σ*) of the methyl group into the σ* orbital on C–C in o-carborane could decrease the electron-withdrawing ability of the o-carborane unit.52 This effect could induce a slightly higher LUMO level followed by a larger band gap, and it is implied that the observation of the emission band in the shorter wavelength region compared to that of CBH could be induced (Table S4, ESI†). The calculations for emission wavelengths of the ICT emission (733 nm for CBH and 692 nm for CBMe) also supported this issue. Due to the presence of the methyl group on the adjacent carbon in the o-carborane unit, the molecular conformation is restricted. As a result, limitation to the ICT character can be induced, followed by relatively-blue-shifted emission compared to that of the H-substituted compound, according to the previous report on emission properties of the series of anthracene–o-carborane dyads with various substituents.55 A similar tendency in the emission wavelength was also suggested in this study.
Conclusion
It was demonstrated that the methyl substituent in the o-carborane unit dramatically influenced optical properties including luminescent color, emission intensity and sensitivity to external stimuli and environmental responsiveness in the bis-o-carborane-substituted 1,4-bis(phenylethynyl)benzene system. Basically, these changes can be explained by the degree of molecular motions and structures determined by the substituents. As a consequence, a variety of useful functions for developing advanced optical materials were obtained. Furthermore, according to computer calculation data, it was implied that dual o-carborane units might individually play different roles from each other in the electronic conjugation in the excited state. This speculation could be valid for establishing design strategies not only to precisely control emission efficiencies by external stimuli and environmental factors but also to increase the diversity of luminescence chromic behaviors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by The Kyoto Technoscience Center (for K. T.) and a Grant-in-Aid for Scientific Research on Innovative Areas “New Polymeric Materials Based on Element-Blocks (No. 2401)” (JSPS KAKENHI Grant Number JP24102013).References
- Y. Chujo and K. Tanaka, Bull. Chem. Soc. Jpn., 2015, 88, 633–643 CrossRef CAS.
- M. Gon, K. Tanaka and Y. Chujo, Polym. J., 2018, 50, 109–126 CrossRef.
- K. Tanaka and Y. Chujo, NPG Asia Mater., 2015, 7, e223 CrossRef CAS.
- H. Yamane, S. Ohtani, K. Tanaka and Y. Chujo, Tetrahedron Lett., 2017, 58, 2989–2992 CrossRef CAS.
- H. Yamane, K. Tanaka and Y. Chujo, Tetrahedron Lett., 2015, 56, 6786–6790 CrossRef CAS.
- K. Suenaga, K. Tanaka and Y. Chujo, Chem. – Eur. J., 2017, 23, 1409–1414 CrossRef CAS PubMed.
- M. Yamaguchi, S. Ito, A. Hirose, K. Tanaka and Y. Chujo, J. Mater. Chem. C, 2016, 3, 5314–5319 RSC.
- M. Yamaguchi, S. Ito, A. Hirose, K. Tanaka and Y. Chujo, Mater. Chem. Front., 2017, 1, 1573–1579 RSC.
- K. Tanaka and Y. Chujo, Macromol. Rapid Commun., 2012, 33, 1235–1255 CrossRef CAS PubMed.
- R. Yoshii, A. Nagai, K. Tanaka and Y. Chujo, Macromol. Rapid Commun., 2014, 35, 1315–1319 CrossRef CAS PubMed.
- K. Tanaka, T. Yanagida, A. Hirose, H. Yamane, R. Yoshii and Y. Chujo, RSC Adv., 2015, 5, 96653–96659 RSC.
- H. Yeo, K. Tanaka and Y. Chujo, Macromolecules, 2016, 49, 8899–8904 CrossRef CAS.
- H. Yamane, S. Ito, K. Tanaka and Y. Chujo, Polym. Chem., 2016, 7, 2799–2807 RSC.
- R. Yoshii, K. Tanaka and Y. Chujo, Macromolecules, 2014, 47, 2268–2278 CrossRef CAS.
- S. Ito, A. Hirose, M. Yamaguchi, K. Tanaka and Y. Chujo, Polymers, 2017, 9, 68–78 CrossRef.
- A. Hirose, K. Tanaka, R. Yoshii and Y. Chujo, Polym. Chem., 2015, 6, 5590–5595 RSC.
- K. Suenaga, R. Yoshii, K. Tanaka and Y. Chujo, Macromol. Chem. Phys., 2016, 217, 414–417 CrossRef CAS.
- R. Yoshii, K. Suenaga, K. Tanaka and Y. Chujo, Chem. – Eur. J., 2015, 21, 7231–7237 CrossRef CAS PubMed.
- K. Suenaga, K. Tanaka and Y. Chujo, Eur. J. Org. Chem., 2017, 5191–5196 CrossRef CAS.
- S. Ohtani, M. Gon, K. Tanaka and Y. Chujo, Chem. – Eur. J., 2017, 23, 11827–11833 CrossRef CAS PubMed.
- T. Matsumoto, H. Takamine, K. Tanaka and Y. Chujo, Mater. Chem. Front., 2017, 1, 2368–2375 RSC.
- K. Suenaga, K. Tanaka and Y. Chujo, Chem. – Eur. J., 2017, 23, 1409–1414 CrossRef CAS PubMed.
- S. Ito, A. Hirose, M. Yamaguchi, K. Tanaka and Y. Chujo, J. Mater. Chem. C, 2016, 3, 5564–5571 RSC.
- V. I. Bregadze, Chem. Rev., 1992, 92, 209–223 CrossRef CAS.
- M. Scholz and E. Hey-Hawkins, Chem. Rev., 2011, 111, 7035–7062 CrossRef CAS PubMed.
- F. Issa, M. Kassiou and L. M. Rendina, Chem. Rev., 2011, 111, 5701–5722 CrossRef CAS PubMed.
- R. Núñez, I. Romero, F. Teixidor and C. Viñas, Chem. Soc. Rev., 2016, 45, 5147–5173 RSC.
- R. Núñez, M. Terrés, A. Ferrer-Ugalde, F. F. D. Biani and F. Teixidor, Chem. Rev., 2016, 116, 14307–14378 CrossRef PubMed.
- R. N. Grimes, Carboranes, Academic Press, Amsterdam, 2nd edn, 2011, pp. 301–540 Search PubMed.
- K. Tanaka, K. Nishino, S. Ito, H. Yamane, K. Suenaga, K. Hashimoto and Y. Chujo, Faraday Discuss., 2017, 196, 31–42 RSC.
- K. Nishino, K. Hashimoto, K. Tanaka, Y. Morisaki and Y. Chujo, Tetrahedron Lett., 2016, 57, 2025–2028 CrossRef CAS.
- K. Nishino, Y. Morisaki, K. Tanaka and Y. Chujo, New J. Chem., 2017, 15, 10550–10554 RSC.
- X. Li, H. Yan and Q. Zhao, Chem. – Eur. J., 2016, 22, 1888–1898 CrossRef CAS PubMed.
- S. Mukherjee and P. Thilagar, Chem. Commun., 2016, 52, 1070–1093 RSC.
- L. Böhling, A. Brockhinke, J. Kahlert, L. Weber, R. A. Harder, D. S. Yufit, J. A. K. Howard, J. A. H. MacBride and M. A. Fox, Eur. J. Inorg. Chem., 2016, 403–412 CrossRef.
- L. Weber, J. Kahlert, R. Brockhinke, L. Böhling, J. Halama, A. Brockhinke, H.-G. Stammler, B. Neumann, C. Nervi, R. A. Harder and M. A. Fox, Dalton Trans., 2013, 42, 10982–10996 RSC.
- J. Kahlert, L. Böhling, A. Brockhinke, H.-G. Stammler, B. Neumann, L. M. Rendina, P. J. Low, L. Weber and M. A. Fox, Dalton Trans., 2015, 44, 9766–9781 RSC.
- Y.-J. Cho, S.-Y. Kim, M. Cho, W.-S. Han, H.-J. Son, D. W. Cho and S. O. Kang, Phys. Chem. Chem. Phys., 2016, 19, 9702–9708 RSC.
- S.-Y. Kim, Y.-J. Cho, G. F. Jin, W.-S. Han, H.-J. Son, D. W. Cho and S. O. Kang, Phys. Chem. Chem. Phys., 2015, 17, 15679–15682 RSC.
- K.-R. Wee, Y.-J. Cho, S. Jeong, S. Kwon, J.-D. Lee, I.-H. Suh and S. O. Kang, J. Am. Chem. Soc., 2012, 134, 17982–17990 CrossRef CAS PubMed.
- B. H. Choi, J. H. Lee, H. Hwang, K. M. Lee and M. H. Park, Organometallics, 2016, 35, 1771–1777 CrossRef CAS.
- R. Furue, T. Nishimoto, I. S. Park, J. Lee and T. Yasuda, Angew. Chem., Int. Ed., 2016, 55, 7171–7175 CrossRef CAS PubMed.
- M. Uebe, A. Ito, Y. Kameoka, T. Sato and K. Tanaka, Chem. Phys. Lett., 2015, 633, 190–194 CrossRef CAS.
- Y. Kameoka, M. Uebe, A. Ito, T. Sato and K. Tanaka, Chem. Phys. Lett., 2014, 615, 44–49 CrossRef CAS.
- M. Eo, M. H. Park, T. Kim, Y. Do and M. H. Lee, Polymer, 2013, 54, 6321–6328 CrossRef CAS.
- T. Kim, H. Kim, K. M. Lee, Y. S. Lee and M. H. Lee, Inorg. Chem., 2013, 52, 160–168 CrossRef CAS PubMed.
- A. Ferrer-Ugalde, J. Cabrera-González, E. J. Juárez-Pérez, F. Teixidor, E. Pérez-Inestrosa, J. M. Montenegro, R. Sillanpää, M. Haukka and R. Núñez, Dalton Trans., 2017, 46, 2091–2104 RSC.
- J. J. Peterson, A. R. Davis, M. Were, E. B. Coughlin and K. R. Carter, ACS Appl. Mater. Interfaces, 2011, 3, 1796–1799 CAS.
- D. Tu, P. Leong, Z. Li, R. Hu, C. Shi, K. Y. Zhang, H. Yan and Q. Zhao, Chem. Commun., 2016, 52, 12494–12497 RSC.
- W. Zhang, Y. Luo, Y. Xu, L. Tian, M. Li, R. He and W. Shen, Dalton Trans., 2015, 44, 18130–18137 RSC.
- L. Zhu, X. Tang, Q. Yu, W. Lv, H. Yan, Q. Zhao and W. Huang, Chem. – Eur. J., 2015, 21, 4721–4730 CrossRef CAS PubMed.
- H. Naito, K. Nishino, Y. Morisaki, K. Tanaka and Y. Chujo, Angew. Chem., Int. Ed., 2017, 56, 254–259 CrossRef CAS PubMed.
- R. Núñez, P. Farràs, F. Teixidor, C. Viñas, R. Sillanpää and R. Kivekäs, Angew. Chem., Int. Ed., 2006, 45, 1270–1272 CrossRef PubMed.
- K. Nishino, H. Yamamoto, K. Tanaka and Y. Chujo, Org. Lett., 2016, 18, 4064–4067 CrossRef CAS PubMed.
- H. Naito, K. Nishino, Y. Morisaki, K. Tanaka and Y. Chujo, Chem. – Asian J., 2017, 12, 2134–2138 CrossRef CAS PubMed.
- H. Naito, K. Nishino, Y. Morisaki, K. Tanaka and Y. Chujo, J. Mater. Chem. C, 2017, 4, 10047–10054 RSC.
- J. Cabrera-González, S. Bhattacharyya, B. Milián-Medina, F. Teixidor, N. Farfán, R. Arcos-Ramos, V. Vargas-Reyes, J. Gierschner and R. Núñez, Eur. J. Inorg. Chem., 2017, 4575–4580 CrossRef.
- D. Tu, P. Leong, S. Guo, H. Yan, C. Lu and Q. Zhao, Angew. Chem., Int. Ed., 2017, 56, 11370–11374 CrossRef CAS PubMed.
- L. Weber, J. Kahlert, R. Brockhinke, L. Böhling, A. Brockhinke, H.-G. Stammler, B. Neumann, R. A. Harder and M. A. Fox, Chem. – Eur. J., 2012, 18, 8347–8357 CrossRef CAS PubMed.
- H. Naito, Y. Morisaki and Y. Chujo, Angew. Chem., Int. Ed., 2015, 54, 5084–5087 CrossRef CAS PubMed.
- K. Nishino, H. Yamamoto, K. Tanaka and Y. Chujo, Asian J. Org. Chem., 2017, 6, 1818–1822 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1581419. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qm00486a |
| ‡ Present address: Department of Applied Chemistry for Environment, School of Science and Technology, Kwansei Gakuin University, 2-1 Gakuen, Sanda, Hyogo 669-1337, Japan. |
| This journal is © the Partner Organisations 2018 |