
A large-bandgap small-molecule electron acceptor utilizing a new indacenodibenzothiophene core for organic solar cells†
Pengfei
Wang‡
ab,
Haijun
Fan‡
 a,
Cheng
Zhang
ab and
Xiaozhang
Zhu
*ab
a,
Cheng
Zhang
ab and
Xiaozhang
Zhu
*ab
aBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Organic Solids, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, China. E-mail: xzzhu@iccas.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, 100049, China
First published on 14th November 2017
Abstract
A weak electron-donating ladder-type heteroarene, indacenodibenzothiophene (IDBT), is designed and synthesized for the development of acceptor–donor–acceptor (A–D–A)-type small-molecule acceptors featuring a large optical bandgap to match high-performance low-bandgap electron donors. Small-molecule acceptor NIDBT based on the IDBT core and 2-(2,3-dihydro-3-oxo-1H-inden-1-ylidene)propanedinitrile as an electron-deficient end group is designed and synthesized. NIDBT shows a wide optical bandgap of 1.84 eV with a high absorption coefficient. A low-bandgap polymer PTB7-Th is selected as the donor to construct photovoltaic devices with NIDBT as the acceptor. After a thermal-annealing treatment at 160 °C for 10 min, an optimal power conversion efficiency (PCE) of 4.45% with an open-circult voltage (Voc) of 0.831 V, a short-circuit current (Jsc) of 11.45 mA cm−2 and a fill factor (FF) of 46.73% is realized.
Introduction
Solution-processed bulk-heterojunction (BHJ) organic solar cells (OPVs) have drawn increasing attention due to their desirable properties such as low-cost, light weight and mechanism flexibility.1–5 Recently, non-fullerene small-molecule electron acceptors (NFAs) have emerged as promising alternatives to fullerene derivatives in OPVs attributed to their broad and strong absorption with adjustable energy levels and feasible synthesis.6–8 Various NFAs have been reported based on diverse π-conjugated frameworks such as perylene diimide (PDI),9–12 naphthalene diimide (NDI),13–15 diketopyrrolopyrrole (DPP),16–18 indacenodithiophene (IDT),19–21 indacenodithieno[3,2-b]thiophene (IDTT),22–24etc. Among these, NFAs with an acceptor–donor–acceptor (A–D–A) arrangement appear to be the most effective for constructing highly efficient OPV devices. Zhan et al. reported a series of NFAs including ITIC, ITIC-Th, IDIC, etc. utilizing indacenodithiophene (IDT) or indacenodithieno[3,2-b]thiophene (IDTT) as a central core with large steric hindrance and electron-rich properties, leading to good photovoltaic performance.7,21–23 By modifying the end groups of ITIC, Hou et al. developed a novel acceptor ITCC with thienyl-fused indanone end-groups and a PCE of 11.4% with an impressive Voc of 1.01 V was obtained.25 Li et al. prepared an acceptor m-ITIC by executing the modification on the aromatic side chain of ITIC, exhibiting a PCE of 11.77% by matching a medium bandgap donor J61.26 Recently, electron acceptors employing benzo[1,2-b:4,5-b′]dithiophene (BDT) as a building block were developed, and good efficiencies were achieved.27–30 By rational selection of the electron donor and systematic device optimization, PCEs in excess of 12% have been achieved.23,31,32To date, most A–D–A type NFAs show low optical bandgaps, which can match large bandgap polymer donors such as PBDB-T,33 J series,34,35 FTAZ,36etc. Considering the fact that many high-performance low-bandgap donor materials have been developed during the last decades for OPVs applying fullerene acceptors, we think that the development of large-bandgap NFAs to match low-bandgap donors should also be a very promising route to achieve high-performance OPVs. For example, Peng et al. reported a PCE of 10.26% based on a large-bandgap acceptor SFBRCN and a low-bandgap donor PTB7-Th.37 As the intramolecular charge-transfer (ICT) property plays a decisive role in the absorption property of A–D–A-type NFAs, in order to design NFAs with large optical bandgaps, we need to weaken the ICT property by either using weak electron-donating core or weak electron-accepting moieties. Accordingly, NFAs with a weak electron-rich fluorene core normally show quite large optical bandgaps. For example, McCulloch et al. reported a novel rhodamine flanked acceptor FBR with a large optical bandgap of 2.14 eV, fabricating the device with a donor P3HT to deliver a PCE of 4.11%.38 Chen et al. developed a large-bandgap acceptor DICTF, employing PTB7-Th as a donor to afford a PCE of 7.93%.39
Electron-rich IDTT is one of the most important building blocks for the design of low-bandgap NFAs. Considering the stronger aromaticity and weaker electron-donating ability of benzene over the thiophene ring, we can reduce the electron-donating ability of IDTT by selectively replacing two of the thiophenes with benzene rings, resulting in a new ladder-type heteroarene, indacenodibenzothiophene (IDBT), that is shown in Fig. 1a. According to the theoretical calculations at B3LYP/6-31G** (Fig. S1, ESI†), we find that the highest occupied molecular orbital (HOMO) energy level of IDBT is significantly deeper than that of IDTT, −5.21 vs. −4.93 eV, indicating the weaker electron-donating ability of IDBT. By attaching IDBT with a strong electron-deficient 2-(2,3-dihydro-3-oxo-1H-inden-1-ylidene)propanedinitrile as the terminal end groups, a new electron acceptor NIDBT was designed and synthesized having a planar and rigid molecular A–D–A framework (Fig. 1b). Indeed, NIDBT displays a large optical bandgap of 1.84 eV, covering the absorption region of 450–670 nm. The energy levels of NIDBT are suitable as an electron acceptor. We selected low-bandgap PTB7-Th as the electron donor to match NIDBT and fabricated OPV devices.40 A PCE of 3.62% was obtained for devices with as-cast blend films. After thermal annealing treatment, the OPV devices could deliver a considerably increased PCE of 4.45%.
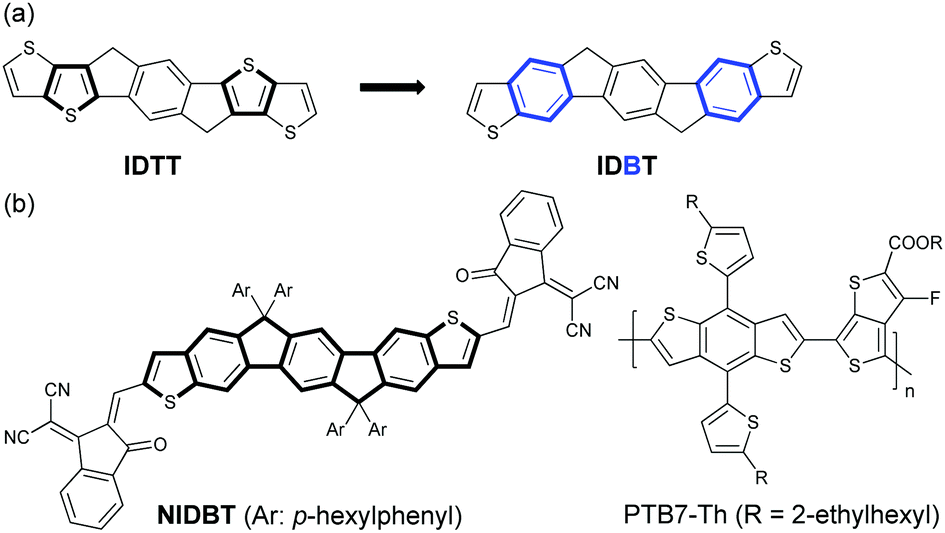 | ||
| Fig. 1 (a) Design of indenofluorenodithiophene (IDBT). (b) Chemical structure of NIDBT and polymer donor PTB7-Th. | ||
Results and discussion
Material preparation
The synthetic route for NIDBT is shown in Scheme 1. Firstly, Suzuki coupling reaction of diethyl 2,5-dibromoterephthalate and (3-chloro-4-methoxyphenyl)boronic acid led to the formation of compound 1 in 93% yield. The double nucleophilic addition reaction between the ester groups of compound 1 and the freshly prepared (4-hexylphenyl)magnesium bromide afforded tertiary alcohol that was used for intramolecular cyclization to give a crucial intermediate 2 in 60% yield for two steps. Demethylation of compound 2 with BBr3 generated compound 3 in 96% yield. Compound 4 was synthesized from compound 3 and trifluoromethanesulfonic anhydride in 97% yield. Then compound 5 was obtained by Pd-catalyzed Sonogashira coupling reaction in 73% yield. Compound 5 gives compound IDBT of 87% yield via intramolecular cyclization reaction using sodium sulfide nonahydrate at 190 °C. Compound IDBT was treated with n-butyl lithium in anhydrous THF at −78 °C, which was followed by reaction with dry DMF to afford dialdehyde IDBT-CHO in 84% yield. Finally, NIDBT was synthesized through Knoevenagel condensation reaction between IDBT-CHO and 2-(2,3-dihydro-3-oxo-1H-inden-1-ylidene)propanedinitrile (INCN) in 45% yield as a black solid. NIDBT exhibits good solubility in common solvents such as dichloromethane, chloroform and chlorobenzene. The thermal stability of NIDBT was investigated by thermogravimetric analysis (TGA) (Fig. S2, ESI†) with a decomposition temperature of 373.5 °C (5% weight loss), indicating a good thermal stability. According to the differential scanning calorimetry curve (Fig. S3, ESI†), a remarkable melting peak at 286 °C was observed.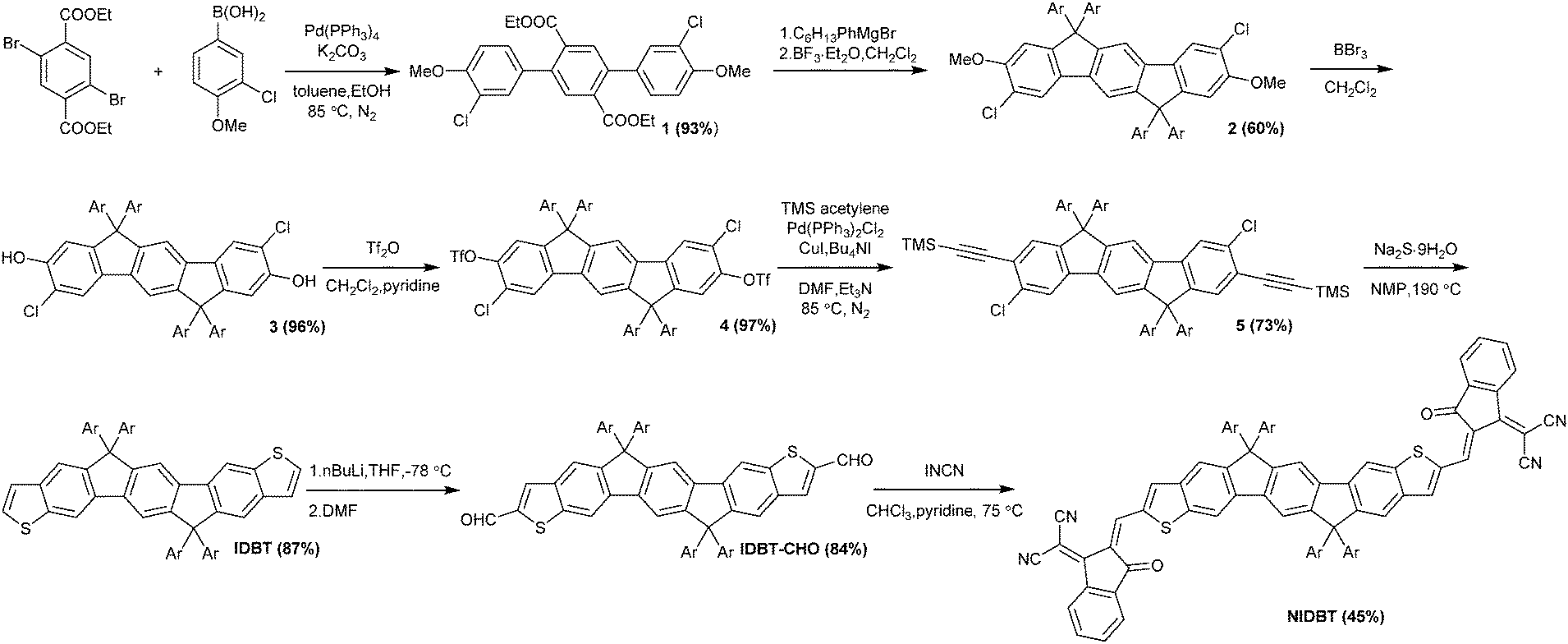 | ||
| Scheme 1 Synthetic procedures for NIDBT. | ||
Photophysical and electrochemical properties
The ultraviolet-visible (UV-vis) absorption spectra of compound NIDBT were presented in Fig. 2a. NIDBT in dilute chloroform solution exhibits a strong absorption with a peak at 593 nm and a high maximum extinction coefficient of 1.51 × 105 M−1 cm−1. From solution to film, a maximum absorption of 605 nm indicates an obvious bathochromic shift, implying that NIDBT has strong intermolecular π–π interactions in the solid state. The optical bandgap of NIDBT was calculated to be 1.84 eV according to the absorption onset of the film. Cyclic voltammetry (CV) was employed to estimate the energy level of NIDBT as presented in Fig. 2b. The HOMO and lowest unoccupied molecular orbital (LUMO) energy levels were estimated to be −5.87 and −3.91 eV, respectively. By contrast, ITIC with a IDTT core shows an obvious redshifted absorption with a much narrower optical bandgap of 1.59 eV,7 originating from stronger intramolecular charge-transfer. The detailed photoelectric data are summarized in Table 1.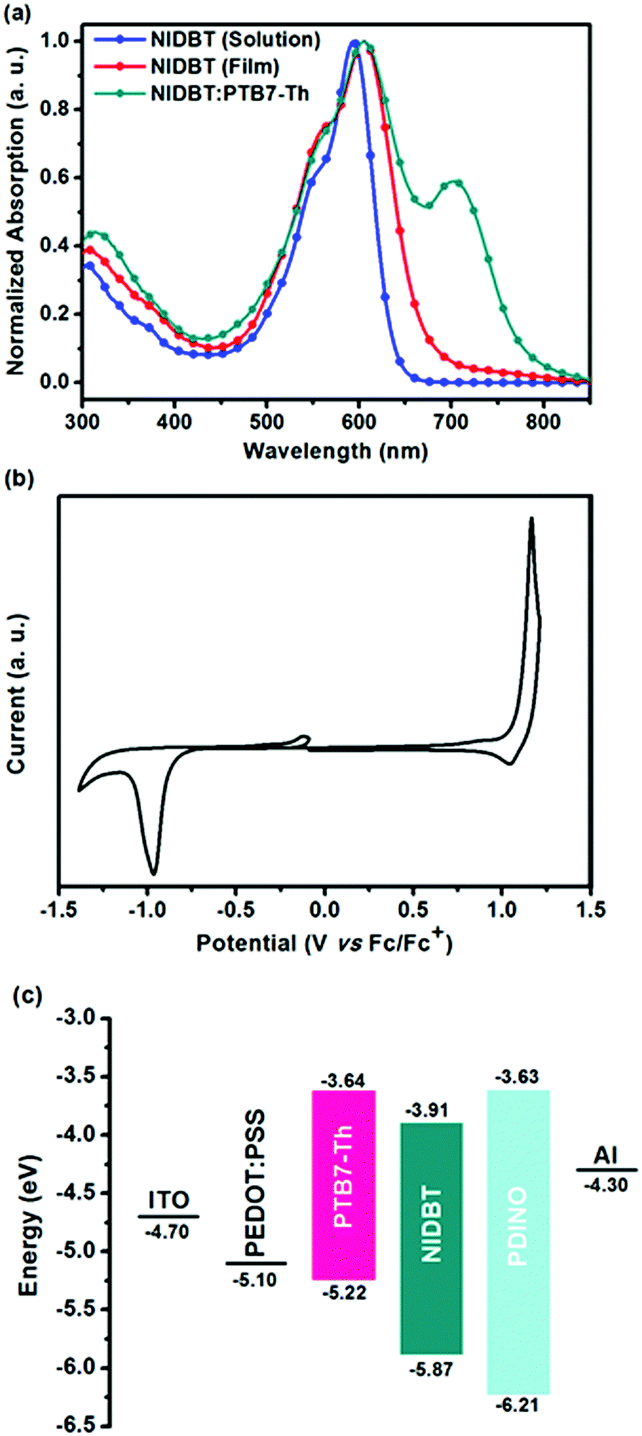 | ||
| Fig. 2 (a) Normalized UV-vis absorption spectra of NIDBT in chloroform, thin films and blend films spin-coated from chloroform solution. (b) Cyclic voltammogram of NIDBT. (c) Energy diagram of the components in OPVs. | ||
| Acceptors |
λ
absmax
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (sol., nm)
(sol., nm) |
ε (×105 M−1 cm−1) |
λ
absmax
(film, nm) |
λ absonset (film, nm) |
E
optg
(eV) |
E HOMO (eV) | E LUMO (eV) |
E
CVg
(eV) |
|---|---|---|---|---|---|---|---|---|
| a Absorption maximum in solution. b Molar extinction coefficient at λmax in solution. c Absorption maximum in a film. d E optg = 1240/λonset. e HOMO and LUMO energy levels were estimated from the onsets of oxidation and reduction processes. f E CVg = ELUMO − EHOMO. g The data of ITIC are from ref. 7. | ||||||||
| NIDBT | 593 | 1.51 | 605 | 673 | 1.84 | −5.87 | −3.91 | 1.96 |
| ITIC | 664 | 1.3 | 702 | 780 | 1.59 | −5.48 | −3.83 | 1.65 |
Photovoltaic properties and charge transport
To investigate the photovoltaic property of NIDBT as the electron acceptor in OSCs, solution-processed devices with the structure of ITO/PEDOT:PSS (poly(3,4-ethylenedioxythiopene):poly(styrenesulfonate))/PTB7-Th:NIDBT/PDINO/Al using PTB7-Th as an electron donor were fabricated, whereas PDINO as a cathode interlayer was reported by Li et al.41 The device with the as-cast film from chlorobenzene gave a PCE of 3.62% with an open-circult voltage (Voc) of 0.808 V, a short-circuit current (Jsc) of 11.33 mA cm−2 and a fill factor (FF) of 39.53%. To improve the photovoltaic properties of the device, thermal annealing treatment was carried out. It was found that when thermal annealing at 160 °C for 10 min was performed, a PCE of 4.45% with a remarkably increased FF of 46.73% was obtained. The current density–voltage (J–V) curves of the device with TA and without TA under 100 mW cm−2 AM 1.5G illumination are shown in Fig. 3a, and their corresponding photovoltaic parameters are shown in Table 2.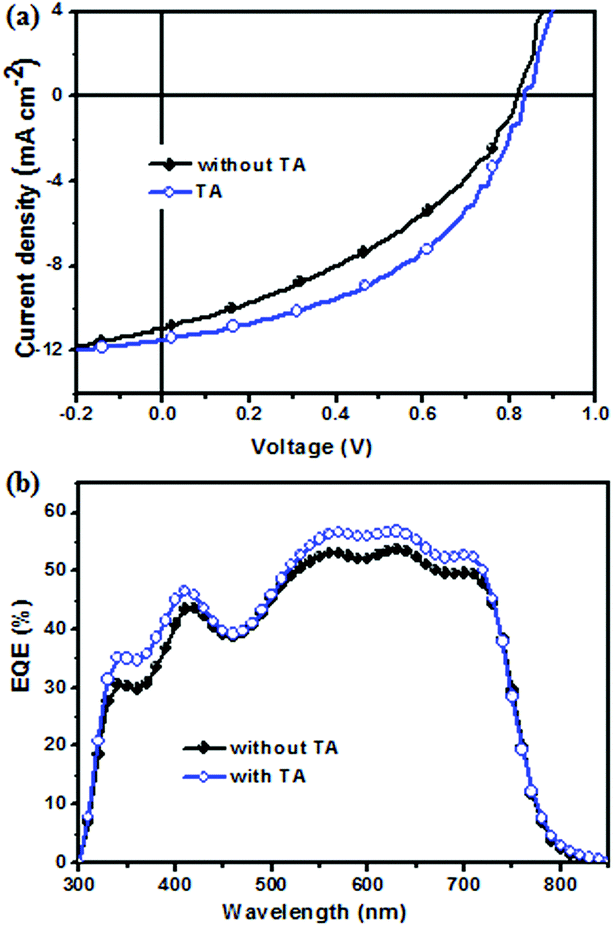 | ||
| Fig. 3 (a) J–V curves and (b) EQE curves of the corresponding devices. | ||
The external quantum efficiency (EQE) curve of the device with TA and without TA treatment is displayed in Fig. 3b. The Jsc value of the device with TA was calculated to be 11.85 mA cm−2 from the EQE spectra, which was in good agreement with the photovoltaic J–V curve test (∼3% error). In contrast to the device without TA, the device with TA had a higher EQE value in the range of 330–720 nm, reaching a maximal EQE value of 56%, which indicated that TA was beneficial to improve the Jsc of the device.
The charge transport properties of the blend film without TA and with TA were detected by the space-charge-limited current (SCLC) method with the device structures of ITO/PEDOT:PSS/active layer/Au for hole mobility and Al/active layer/Al for electron mobility (the detailed data are shown in Fig. S4 and Table S1, ESI†). The hole and electron mobility values of NIDBT in blend films without TA were 7.2 × 10−4 and 3.55 × 10−5 cm2 V−1 S−1, respectively. After TA treatment, the hole and electron mobility values were changed to be 3.8 × 10−4 cm2 V−1 S−1, and 6.87 × 10−5 cm2 V−1 S−1, respectively. The ratio value of μh/μe without TA was calculated to be 20.2. After the TA treatment, the ratio value of μh/μe was reduced to be 5.53. The electron mobility as well as more balanced charge transport benefits the improvement of Jsc and FF.
Morphology characterization
To study the relationship between the morphology of the blend film and the device performance, atomic force microscopy (AFM) was performed. In Fig. 4, the as-cast film exhibits a surface featuring many small but dense aggregates with a root mean-square roughness (RMS) of 1.55 nm.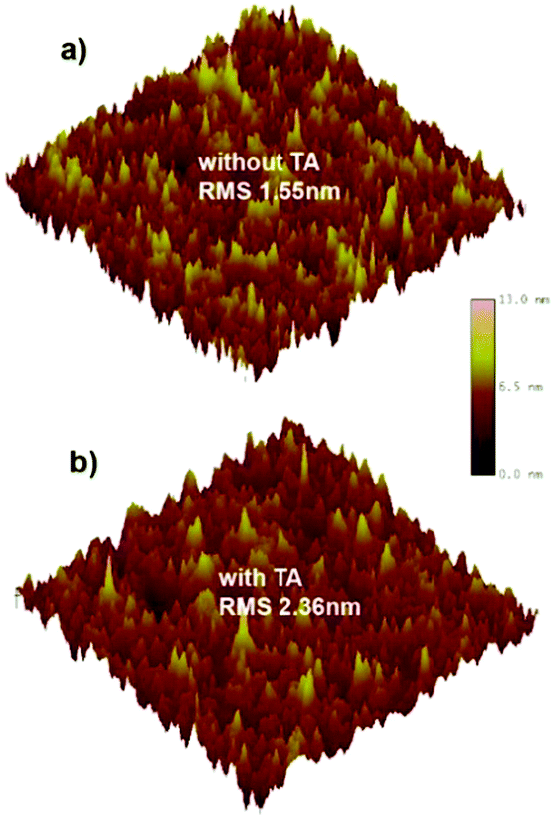 | ||
| Fig. 4 AFM height image of the blend film (a) without TA and (b) with TA. | ||
After TA treatment, the film shows more large but loose aggregates with an increased RMS of 2.36 nm. Such size-increase of the aggregation may relate to the enhanced crystallinity of NIDBT, which tends to corrupt the continuity of the polymer donor phase and thus reduces the hole mobility but helps to increase the electron mobility. However, accompanying such adverse trends in the change of the hole and electron mobility, a more balanced charge transport is realized, which contributes to the higher FF by reducing the recombination.
Conclusions
In summary, we designed and synthesized a new heteroarene with weak electron-donating ability, IDBT, for application in fullerene-free organic solar cells. The A–D–A type non-fullerene small-molecule acceptor NIDBT based on an IDBT core shows a large optical bandgap of 1.84 eV. OPV devices were fabricated by choosing PTB7-Th as the low-bandgap donor, leading to a PCE of 4.45% with a Voc of 0.831 V, Jsc of 11.45 mA cm−2 and FF of 46.73% by TA treatment. Although the PCE is relatively low at the present stage, new NFAs with IDBT cores may deliver high OPV performance via proper molecular design, which will be reported in due course.Experimental procedure
Diethyl 3,3′′-dichloro-4,4′′-dimethoxy-[1,1′:4′,1′′-terphenyl]-2′,5′-dicarboxylate (1)
A mixture of diethyl 2,5-dibromoterephthalate (1.900 g, 5 mmol), (3-chloro-4-methoxyphenyl)boronic acid (1.957 g, 2.1 equiv.), Pd(PPh3)4 (288.9 mg, 5 mol%) and K2CO3 (2.073 g, 3 equiv.) in toluene (20 mL) was degassed with nitrogen for 10 min and then ethanol (5 mL) was added. The mixture was stirred at 85 °C for one day. The reaction mixture was cooled to room temperature and was diluted with dichloromethane and washed with water. The organic phase was separated and the aqueous phase was extracted with dichloromethane. The combined organic phase was dried over anhydrous MgSO4 and concentrated under reduced pressure. The residue was purified by silica-gel column chromatography to give the desired product 1 as a white solid (2.340 g, 93% yield). 1H NMR (400 MHz, CDCl3): δ 7.76 (s, 2H), 7.39 (s, 2H), 7.22 (d, J = 8.3 Hz, 2H), 6.98 (d, J = 8.4 Hz, 2H), 4.18 (q, J = 6.9 Hz, 4H), 3.95 (s, 6H), 1.12 (t, J = 7.0 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ 167.6, 154.7, 139.4, 133.4, 133.2, 131.9, 130.3, 127.8, 122.3, 111.7, 61.4, 56.2, 13.8; HRMS (MALDI) calcd for C26H24Cl2NaO6 [M + Na]+: 525.0842; found: 525.0846.3,9-Dichloro-6,6,12,12-tetrakis(4-hexylphenyl)-2,8-dimethoxy-6,12-dihydroindeno[1,2-b]fluorene (2)
A solution of compound 1 (755.1 mg, 1.5 mmol) in dry THF (10 mL) was added to freshly prepared 4-hexylphenyl-1-magnesium bromide that was prepared by slowly adding 1-bromo-4-hexylbenzene (4.351 g, 12 equiv.) to Mg (481.1 mg, 13.2 equiv.) in dry THF (15 mL), and then was refluxed for 2 h under a nitrogen atmosphere. Then the reaction was refluxed for 16 h and was cooled to room temperature. The mixture was quenched with H2O under an ice bath, and extracted with diethyl ether. The organic phase was dried over anhydrous MgSO4, and concentrated under reduced pressure. The residue in dry dichloromethane (20 mL) was mixed with BF3·Et2O (3 mL) under an ice bath and a nitrogen atmosphere. The mixture was stirred for 1 h at room temperature and was poured into water. The organic phase was separated and dried over anhydrous MgSO4 and concentrated under reduced pressure. The residue was purified by silica-gel column chromatography to give the desired product 2 as a white solid (919.4 mg, 60% yield). 1H NMR (400 MHz, CDCl3): δ 7.62 (s, 2H), 7.56 (s, 2H), 7.13 (d, J = 7.7 Hz, 8H), 7.06 (d, J = 7.7 Hz, 8H), 6.91 (s, 2H), 3.82 (s, 6H), 2.56 (t, J = 7.6 Hz, 8H), 1.65–1.54 (m, 8H), 1.39–1.23 (m, 24H), 0.87 (t, J = 5.4 Hz, 12H); 13C NMR (100 MHz, CDCl3): δ 154.5, 151.9, 151.3, 142.8, 141.5, 138.5, 133.7, 128.3, 128.1, 122.1, 121.6, 117.1, 110.1, 64.8, 56.4, 35.5, 31.7, 31.3, 29.1, 22.6, 14.1; HRMS (MALDI) calcd for C70H80Cl2O2 [M]+: 1022.5530; found: 1022.5538.3,9-Dichloro-6,6,12,12-tetrakis(4-hexylphenyl)-6,12-dihydroindeno[1,2-b]fluorene-2,8-diol (3)
Compound 2 (992.6 mg, 0.969 mmol) in dry dichloromethane (30 mL) was mixed with BBr3 (560 μL, 6 equiv.) under an ice bath and a nitrogen atmosphere, and then was stirred overnight. The reaction mixture was poured into water. The organic phase was separated and dried over anhydrous MgSO4 and concentrated under reduced pressure. The residue was purified by silica-gel column chromatography to give the desired product 3 as a white solid (929.1 mg, 96% yield). 1H NMR (400 MHz, CDCl3): δ 7.57 (d, J = 5.0 Hz, 4H), 7.12 (d, J = 7.7 Hz, 8H), 7.08–7.01 (m, 10H), 5.50 (s, 2H), 2.55 (t, J = 7.7 Hz, 8H), 1.59 (m, 8H), 1.39–1.24 (m, 24H), 0.87 (t, J = 6.0 Hz, 12H); 13C NMR (100 MHz, CDCl3): δ 152.7, 151.2, 150.6, 142.7, 141.5, 138.5, 133.9, 128.3, 128.0, 120.2, 119.4, 117.2, 114.1, 64.5, 35.5, 31.7, 31.3, 29.1, 22.6, 14.1; HRMS (MALDI) calcd for C68H76Cl2O2 [M]+: 994.5217; found: 994.5217.3,9-Dichloro-6,6,12,12-tetrakis(4-hexylphenyl)-6,12-dihydroindeno[1,2-b]fluorene-2,8-diyl bis(trifluoromethanesulfonate) (4)
Compound 3 (868.0 mg, 0.8713 mmol) in dry dichloromethane (50 mL) and super dry pyridine (2 mL) were added to trifluoromethanesulfonic anhydride (440 μL, 3 equiv.) under an ice bath and a nitrogen atmosphere. The reaction mixture was then stirred for 3 h at room temperature. After the reaction completion, the reaction mixture was poured into H2O. The organic phase was separated and dried over anhydrous MgSO4 and concentrated under reduced pressure. The residue was purified by silica-gel column chromatography to give the desired product 4 as a white solid (1.065 g, 97% yield). 1H NMR (400 MHz, CDCl3): δ 7.78 (s, 2H), 7.71 (s, 2H), 7.31 (s, 2H), 7.08 (s, 16H), 2.56 (t, J = 7.7 Hz, 8H), 1.57 (m, 8H), 1.38–1.24 (m, 24H), 0.87 (t, J = 5.2 Hz, 12H); 13C NMR (100 MHz, CDCl3): δ 152.5, 152.3, 144.6, 142.3, 141.3, 140.7, 138.5, 128.7, 127.7, 126.7, 122.5, 121.0, 118.63 (q, J = 320.8 Hz), 118.61, 64.9, 35.5, 31.7, 31.3, 29.1, 22.6, 14.1; HRMS (APCI) calcd for C70H75O6Cl2F6S2 [M + H]+: 1259.4281; found: 1259.4265.((3,9-Dichloro-6,6,12,12-tetrakis(4-hexylphenyl)-6,12-dihydroindeno[1,2-b]fluorene-2,8-diyl)bis(ethyne-2,1-diyl))bis(trimethylsilane) (5)
A mixture of compound 4 (1.012 g, 0.803 mmol), bis(triphenylphosphine)palladium(II) chloride (67.6 mg, 12 mol%), copper iodide (45.9 mg, 30 mol%), tetrabutylammonium iodide (889.8 mg, 3 equiv.) in N,N-dimethylformamide (12 mL) and triethylamine (2.4 mL) was degassed with nitrogen for 10 min, to which ethynyltrimethylsilane (340 μL, 3 equiv.) was added. The reaction mixture was stirred at 75 °C overnight. After the reaction completion, the mixture was cooled to room temperature. The reaction mixture was diluted with dichloromethane and was washed with water. The organic phase was separated and was dried over anhydrous MgSO4 and concentrated under reduced pressure. The residue was purified by silica-gel column chromatography to give the desired product 5 as a yellow solid (680.8 mg, 73% yield). 1H NMR (400 MHz, CDCl3): δ 7.65 (d, J = 2.1 Hz, 4H), 7.47 (s, 2H), 7.08 (q, J = 8.4 Hz, 16H), 2.61–2.51 (m, 8H), 1.59 (m, 8H), 1.39–1.24 (m, 24H), 0.88 (t, J = 6.7 Hz, 12H), 0.25 (s, 18H); 13C NMR (100 MHz, CDCl3): δ 152.5, 150.2, 142.0, 141.7, 141.5, 139.3, 135.8, 130.9, 128.4, 128.0, 121.5, 120.9, 118.3, 102.2, 100.5, 64.4, 35.5, 31.7, 31.3, 29.1, 22.6, 14.1, −0.1; HRMS (MALDI) calcd for C78H92Cl2Si2 [M]+: 1154.6109; found: 1154.6113.IDBT
A solution of sodium sulfide nonahydrate (384.3 mg, 4 equiv.) in 1-methyl-2-pyrrolidinone (8 mL) was degassed with nitrogen for 10 min, to which compound 5 (462.7 mg, 0.4 mmol) was added. The reaction mixture was stirred for 12 h at 190 °C. The reaction mixture was cooled to room temperature, poured into H2O and extracted with dichloromethane. The combined organic phase was dried over anhydrous MgSO4 and concentrated under reduced pressure. The residue was purified by silica-gel column chromatography to give the desired product IDBT as a yellow solid (348.9 mg, 87% yield). 1H NMR (400 MHz, CD2Cl2): δ 8.15 (s, 2H), 7.82 (s, 2H), 7.75 (s, 2H), 7.40 (d, J = 5.2 Hz, 2H), 7.25 (d, J = 5.3 Hz, 2H), 7.20 (d, J = 7.6 Hz, 8H), 7.06 (d, J = 7.6 Hz, 8H), 2.54 (t, J = 7.7 Hz, 8H), 1.60–1.51 (m, 8H), 1.30 (m, 24H), 0.84 (t, J = 5.9 Hz, 12H); 13C NMR (100 MHz, CDCl3): δ 151.9, 149.3, 143.8, 141.2, 139.8, 139.40, 139.35, 137.7, 128.2, 126.2, 124.1, 121.2, 117.9, 113.6, 64.1, 35.6, 31.7, 31.3, 29.2, 22.6, 14.1; HRMS (MALDI) calcd for C72H78S2 [M]+: 1006.5539; found: 1006.5540.IDBT-CHO
A solution of compound IDBT (161.2 mg, 0.16 mmol) in dry THF (8 mL) at −78 °C was slowly added to n-butyllithium in hexane (0.25 mL, 1.6 M, 2.5 equiv.) under a nitrogen atmosphere. The reaction mixture was stirred at −78 °C for 1.5 h and then super dry DMF (37 μL, 3 equiv.) was added. The reaction mixture was stirred at −78 °C for 1 h and then was stirred at room temperature for 1 h, and then was quenched with water. The mixture was extracted with dichloromethane, dried over anhydrous MgSO4 and concentrated under reduced pressure. The residue was purified by silica-gel column chromatography to give the desired product IDBT-CHO as a yellow solid (142.2 mg, 84% yield). 1H NMR (400 MHz, CDCl3): δ 10.04 (s, 2H), 8.17 (s, 2H), 7.93 (s, 2H), 7.89 (s, 2H), 7.86 (s, 2H), 7.19 (d, J = 7.7 Hz, 8H), 7.08 (d, J = 7.9 Hz, 8H), 2.56 (t, J = 7.8 Hz, 8H), 1.58 (m, 8H), 1.38–1.23 (m, 24H), 0.86 (t, J = 6.3 Hz, 12H); 13C NMR (100 MHz, CDCl3): δ 184.3, 152.6, 150.5, 143.2, 143.0, 142.7, 141.7, 140.0, 138.4, 134.7, 128.4, 128.1, 123.7, 118.7, 114.5, 64.2, 35.5, 31.7, 31.3, 29.1, 22.6, 14.1; HRMS (MALDI) calcd for C74H78O2S2 [M]+: 1062.5438; found: 1062.5435.NIDBT
A solution of compound IDBT-CHO (75.5 mg, 0.071 mmol) and 2-(2,3-dihydro-3-oxo-1H-inden-1-ylidene)propanedinitrile (137. 9 mg 10 equiv.) in chloroform (12 mL) was degassed for 5 min, to which super dry pyridine (0.2 mL) was added. The reaction mixture was stirred at 75 °C overnight and then cooled to room temperature. The reaction mixture was poured into methanol (200 mL) and then was filtered. The residue was purified by silica-gel column chromatography, and was further purified by recrystallization (CHCl3/n-hexane) to afford NIDBT as a black solid (45.3 mg, 45% yield). 1H NMR (400 MHz, CDCl3): δ 8.95 (s, 2H), 8.72 (d, J = 7.4 Hz, 2H), 8.17 (s, 2H), 8.11 (s, 2H), 7.98 (d, J = 7.0 Hz, 2H), 7.91 (s, 2H), 7.87 (s, 2H), 7.79 (m, 4H), 7.22 (d, J = 7.9 Hz, 8H), 7.10 (d, J = 8.0 Hz, 8H), 2.61–2.53 (m, 8H), 1.65–1.54 (m, 8H), 1.39–1.24 (m, 24H), 0.86 (t, J = 6.3 Hz, 12H); 13C NMR (150 MHz, CDCl3): δ 187.8, 160.2, 153.1, 150.8, 147.6, 142.9, 142.2, 141.8, 140.4, 140.0, 138.7, 138.3, 137.3, 137.0, 135.5, 134.7, 128.5, 128.1, 125.4, 125.2, 124.1, 119.0, 114.3, 114.1, 113.8, 71.0, 64.1, 35.5, 31.7, 31.3, 29.1, 22.6, 14.1; HRMS (MALDI) calcd for C98H86N4O2S2 [M]+: 1414.6187; found: 1414.6178; anal. calcd for C98H86N4O2S2 (%): C, 83.13; H, 6.12; N, 3.96; found: C, 83.12, H, 6.16; N, 3.95.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Basic Research Program of China (973 Program) (No. 2014CB643502) for financial support, the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB12010200), and the National Natural Science Foundation of China (91333113, 21572234, 61505225).References
- Y.-J. Cheng, S.-H. Yang and C. S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS PubMed.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CAS.
- Y. Li, Acc. Chem. Res., 2012, 45, 723–733 CrossRef CAS PubMed.
- G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153–161 CrossRef CAS.
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS PubMed.
- J. E. Anthony, Chem. Mater., 2011, 23, 583–590 CrossRef CAS.
- Y. Lin, J. Wang, Z.-H. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- C. B. Nielsen, S. Holliday, H.-Y. Chen, S. J. Cryer and I. McCulloch, Acc. Chem. Res., 2015, 48, 2803–2812 CrossRef CAS PubMed.
- D. Meng, H. Fu, C. Xiao, X. Meng, T. Winands, W. Ma, W. Wei, B. Fan, L. Huo, N. L. Doltsinis, Y. Li, Y. Sun and Z. Wang, J. Am. Chem. Soc., 2016, 138, 10184–10190 CrossRef CAS PubMed.
- X. Zhang, Z. Lu, L. Ye, C. Zhan, J. Hou, S. Zhang, B. Jiang, Y. Zhao, J. Huang, S. Zhang, Y. Liu, Q. Shi, Y. Liu and J. Yao, Adv. Mater., 2013, 25, 5791–5797 CrossRef CAS PubMed.
- R. Xin, J. Feng, C. Zeng, W. Jiang, L. Zhang, D. Meng, Z. Ren, Z. Wang and S. Yan, ACS Appl. Mater. Interfaces, 2017, 9, 2739–2746 CAS.
- Q. Wu, D. Zhao, A. M. Schneider, W. Chen and L. Yu, J. Am. Chem. Soc., 2016, 138, 7248–7251 CrossRef CAS PubMed.
- J. W. Jung, J. W. Jo, C.-C. Chueh, F. Liu, W. H. Jo, T. P. Russell and A. K.-Y. Jen, Adv. Mater., 2015, 27, 3310–3317 CrossRef CAS PubMed.
- M. J. Sung, M. Huang, S. H. Moon, T. H. Lee, S. Y. Park, J. Y. Kim, S.-K. Kwon, H. Choi and Y.-H. Kim, Sol. Energy, 2017, 150, 90–95 CrossRef CAS.
- D. Srivani, A. Gupta, S. V. Bhosale, A. L. Puyad, W. Xiang, J. Li, R. A. Evans and S. V. Bhosale, Chem. Commun., 2017, 53, 7080–7083 RSC.
- K. Gao, L. Li, T. Lai, L. Xiao, Y. Huang, F. Huang, J. Peng, Y. Cao, F. Liu, T. P. Russell, R. A. J. Janssen and X. Peng, J. Am. Chem. Soc., 2015, 137, 7282–7285 CrossRef CAS PubMed.
- K. Gao, J. Miao, L. Xiao, W. Deng, Y. Kan, T. Liang, C. Wang, F. Huang, J. Peng, Y. Cao, F. Liu, T. P. Russell, H. Wu and X. Peng, Adv. Mater., 2016, 28, 4727–4733 CrossRef CAS PubMed.
- T. Liang, L. Xiao, K. Gao, W. Xu, X. Peng and Y. Cao, ACS Appl. Mater. Interfaces, 2017, 9, 7131–7138 CAS.
- H. Bai, Y. Wang, P. Cheng, J. Wang, Y. Wu, J. Hou and X. Zhan, J. Mater. Chem. A, 2015, 3, 1910–1914 CAS.
- Y. Lin, Z.-G. Zhang, H. Bai, J. Wang, Y. Yao, Y. Li, D. Zhu and X. Zhan, Energy Environ. Sci., 2015, 8, 610–616 CAS.
- Y. Lin, F. Zhao, Y. Wu, K. Chen, Y. Xia, G. Li, S. K. K. Prasad, J. Zhu, L. Huo, H. Bin, Z.-G. Zhang, X. Guo, M. Zhang, Y. Sun, F. Gao, Z. Wei, W. Ma, C. Wang, J. Hodgkiss, Z. Bo, O. Inganäs, Y. Li and X. Zhan, Adv. Mater., 2017, 29, 1604155 CrossRef PubMed.
- Y. Lin, F. Zhao, Q. He, L. Huo, Y. Wu, T. C. Parker, W. Ma, Y. Sun, C. Wang, D. Zhu, A. J. Heeger, S. R. Marder and X. Zhan, J. Am. Chem. Soc., 2016, 138, 4955–4961 CrossRef CAS PubMed.
- F. Zhao, S. Dai, Y. Wu, Q. Zhang, J. Wang, L. Jiang, Q. Ling, Z. Wei, W. Ma, W. You, C. Wang and X. Zhan, Adv. Mater., 2017, 29, 1700144 CrossRef PubMed.
- J. Zhu, S. Li, X. Liu, H. Yao, F. Wang, S. Zhang, M. Sun and J. Hou, J. Mater. Chem. A, 2017, 5, 15175–15182 CAS.
- H. Yao, L. Ye, J. Hou, B. Jang, G. Han, Y. Cui, G. M. Su, C. Wang, B. Gao, R. Yu, H. Zhang, Y. Yi, H. Y. Woo, H. Ade and J. Hou, Adv. Mater., 2017, 29, 1700254 CrossRef PubMed.
- Y. Yang, Z.-G. Zhang, H. Bin, S. Chen, L. Gao, L. Xue, C. Yang and Y. Li, J. Am. Chem. Soc., 2016, 138, 15011–15018 CrossRef CAS PubMed.
- B. Kan, H. Feng, X. Wan, F. Liu, X. Ke, Y. Wang, Y. Wang, H. Zhang, C. Li, J. Hou and Y. Chen, J. Am. Chem. Soc., 2017, 139, 4929–4934 CrossRef CAS PubMed.
- Y. Li, L. Zhong, B. Gautam, H.-J. Bin, J.-D. Lin, F.-P. Wu, Z. Zhang, Z.-Q. Jiang, Z.-G. Zhang, K. Gundogdu, Y. Li and L.-S. Liang, Energy Environ. Sci., 2017, 10, 1610–1620 CAS.
- Y. Li, L. Zhong, J.-D. Lin, F.-P. Wu, H.-J. Bin, Z. Zhang, L. Xu, Z.-Q. Jiang, Z.-Q. Jiang, F. Liu, T. P. Russell, Y. Li, L.-S. Liang and S. R. Forrest, Sol. RRL, 2017, 1, 1700107 CrossRef.
- J. Wang, W. Wang, X. Wang, Y. Wu, Q. Zhang, C. Yan, W. Ma, W. You and X. Zhan, Adv. Mater., 2017, 29, 1702125 CrossRef PubMed.
- S. Li, L. Ye, W. Zhao, S. Zhang, S. Mukherjee, H. Ade and J. Hou, Adv. Mater., 2016, 28, 9423–9429 CrossRef CAS PubMed.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148–7151 CrossRef CAS PubMed.
- J. Zhu, S. Li, X. Liu, H. Yao, F. Wang, S. Zhang, M. Sun and J. Hou, J. Mater. Chem. A, 2017, 5, 15175–15182 CAS.
- H. Bin, Z.-G. Zhang, L. Gao, S. Chen, L. Zhong, L. Xue, C. Yang and Y. Li, J. Am. Chem. Soc., 2016, 138, 4657–4664 CrossRef CAS PubMed.
- H. Bin, L. Gao, Z.-G. Zhang, Y. Yang, Y. Zhang, C. Zhang, S. Chen, L. Xue, C. Yang, M. Xiao and Y. Li, Nat. Commun., 2016, 7, 13651 CrossRef CAS PubMed.
- S. Dai, F. Zhao, Q. Zhang, T.-K. Lau, T. Li, K. Liu, Q. Ling, C. Wang, X. Lu, W. You and X. Zhan, J. Am. Chem. Soc., 2017, 139, 1336–1343 CrossRef CAS PubMed.
- G. Zhang, G. Yang, H. Yan, J.-H. Kim, H. Ade, W. Wu, X. Xu, Y. Duan and Q. Peng, Adv. Mater., 2017, 29, 1606054 CrossRef PubMed.
- S. Holliday, R. S. Ashraf, C. B. Nielsen., M. Kirkus, J. A. Röhr, C.-H. Tan, E. Collado-Fregoso, A.-C. Knall, J. R. Durrant, J. Nelson and I. McCulloch, J. Am. Chem. Soc., 2015, 137, 898–904 CrossRef CAS PubMed.
- M. Li, Y. Liu, W. Ni, F. Liu, H. Feng, Y. Zhang, T. Liu, H. Zhang, X. Wan, B. Kan, Q. Zhang, T. P. Russell and Y. Chen, J. Mater. Chem. A, 2016, 4, 10409–10413 CAS.
- S.-H. Liao, H.-J. Jhuo, Y.-S. Cheng and S.-A. Chen, Adv. Mater., 2013, 25, 4766–4771 CrossRef CAS PubMed.
- Z.-G. Zhang, B. Yi, Z. Jin, D. Chi, Z. Qi, Y. Li and J. Wang, Energy Environ. Sci., 2014, 7, 1966–1973 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Theoretical calculations of IDBT, TGA, DSC and XRD curve of NIDBT, SCLC mobility plot, and NMR spectra of all compounds. See DOI: 10.1039/c7qm00403f |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2018 |