DOI:
10.1039/C7QM00399D
(Research Article)
Mater. Chem. Front., 2018,
2, 559-565
Waterborne polyacrylates with thermally activated delayed fluorescence and two-state phosphorescence†
Received
31st August 2017
, Accepted 10th January 2018
First published on 10th January 2018
Abstract
Purely organic luminescent materials are extensively used in bioimaging, sensing and organic light-emitting diodes (OLEDs). Anthraquinone (AQ) derivatives with N- and O-substitution are incorporated into waterborne polyacrylates as single-component polymers with both thermally activated delayed fluorescence (TADF) and two-state phosphorescence. Essential structural and optical properties are characterized for these waterborne polyacrylates. With increasing dye loadings, a convergence between singlet and triplet states due to excitonic splittings is observed.

Guoqing Zhang
| Dr Guoqing Zhang is currently a full professor at the Hefei National Laboratory for Physical Sciences at the Microscale at the University of Science and Technology of China (USTC), where he got his Bachelor of Science degree. Prof. Zhang got his PhD degree in Chemistry from the University of Virginia under the supervision of Prof. Cassandra L. Fraser and his research was mostly on the sensing and imaging applications of difluoroboron diketone polylactides. He then proceeded to Harvard to work with Prof. X. Sunny Xie on transposon study in understanding bacterial persistence. In the summer of 2011, Prof. Zhang joined the faculty of USTC where he started to work on the design and synthesis of new phosphorescent materials. His current primary research interests include (1) fluorogenic dyes for assisted surgical procedures, (2) development of methods to achieve highly efficient phosphorescence based on purely organic systems, and (3) the design and synthesis of new antimicrobial polypeptide mimics (APMs). In 2015, Prof. Zhang founded a startup company dedicated to consumer products based on APMs and has successfully raised more than $3 million in venture capitals. In 2016, Prof. Zhang led a team of scientists and artists to win the First Place in National Women's Fashion Design Competition of China based on a class of fractal structures observed during lab research. |
Introduction
Purely organic luminescent materials have attracted a surge of recent interest because of their potential applications in bioimaging,1–3 chemical sensors,4 organic lasers,5 and organic light-emitting diodes (OLEDs).6,7 Kim et al.8,9 developed a new class of RTP luminophores by introducing aromatic carbonyl groups in aromatic systems, with halogen atoms to promote the efficiency of spin–orbit coupling. Fraser et al.10,11 developed a class of single-component, dual-emissive difluoroboron dibenzoylmethane-poly(lactic acid) species with both fluorescence and RTP. Recently, pure OLEDs have been extensively investigated for flat-panel displays and light applications because they are cost efficient, flexible,12,13 and low in toxicity, compared to conventional metal-doped OLEDs. As for conventional fluorescence, the maximal internal electroluminescence (EL) efficiency is 25% because singlet (S1) and triplet (T1) state excitons will be formed in a roughly 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio under electrical excitation according to spin statistics.14 Therefore, the internal EL efficiency of purely luminescent molecules with both fluorescence and RTP may be 100% theoretically. In general, the lifetimes of RTP from purely organic molecules are so long that oxygen quenching and vibrational relaxation could interfere with emissions desirable for OLEDs. A series of thermally activated delayed fluorescence (TADF) materials were developed by Adachi15,16 and others,17,18 where radiative lifetimes could be reduced to the sub-ms domain.
:3 ratio under electrical excitation according to spin statistics.14 Therefore, the internal EL efficiency of purely luminescent molecules with both fluorescence and RTP may be 100% theoretically. In general, the lifetimes of RTP from purely organic molecules are so long that oxygen quenching and vibrational relaxation could interfere with emissions desirable for OLEDs. A series of thermally activated delayed fluorescence (TADF) materials were developed by Adachi15,16 and others,17,18 where radiative lifetimes could be reduced to the sub-ms domain.
Organic molecules with π–π* transitions typically exhibit aggregation-caused quenching (ACQ) due to strong “face-to-face” stacking interactions. In order to circumvent the ACQ effect, Tang19,20 and others21,22 designed a large number of molecules with aggregation-induced emission (AIE) by restricting intramolecular rotation in the solid state. Another way to avoid the ACQ effect is to incorporate small molecules into polymers covalently.2,10,11 Flexible polymeric emitters are suitable for solution-cast films and were successfully applied in OLEDs by Bryce23 and others.24,25
Here we report a different mechanism for TADF in a single-component waterborne polyacrylate system with various ratios of O-substituted anthraquinones (POAQs) and N-substituted AQs (PNAQs) (the synthetic procedure is according to the literature26,27). We found that the generation of TADF for these polymers is very likely due to the fact that they have two distinct emissive triplet states which belong to 3n–π* and 3π–π*, respectively. The energetically close, four-state configuration renders thermal back-population from 3n–π* to 1π–π* and 3π–π* to 1n–π* very effectively, according to the El-Sayed rule. Moreover, when the dye concentration is increased, energy-splitting was observed, which lines up with a recent model we proposed, polymerization-enhanced intersystem crossing (PEX).28 As a result, the luminescent waterborne polyacrylates with both TADF and phosphorescence have the potential to be used for aqueous-based photoluminescence imaging and sensing in biological contexts.
Results and discussion
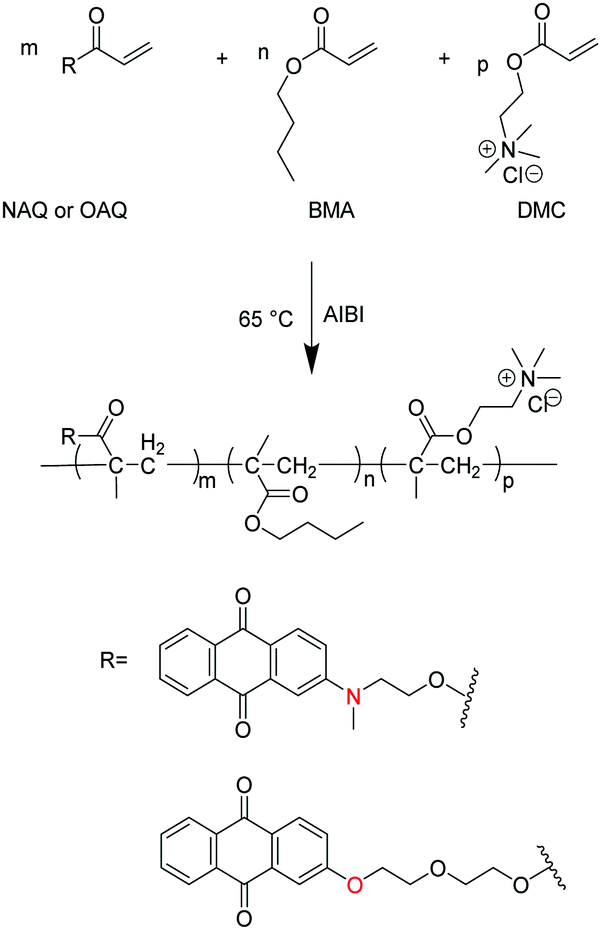
The synthetic procedure of waterborne polyacrylates is shown in Scheme 1, where the random copolymers of PNAQ and POAQ could be obtained via AIBN-initiated emulsion radical polymerization. The molar ratios of NAQ and OAQ can be easily tuned by the amount of dye acrylate monomer fed to the reaction mixture. Thin solid films were obtained by letting the emulsions dry at room temperature on a polytetrafluoroethylene substrate. The FT-IR spectra (Fig. 1 and Fig. S1, ESI†) are employed to analyse the functional groups in PNAQ and POAQ, given that the 1H-NMR spectra are difficult to decipher due to extensive peak overlaps in these types of polymers. Compared to the PNAQ polymer, the characteristic peak at 1720 cm−1 is ascribed to the stretching vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond conjugated to the CC bond. However, the stretching vibration peak of the CO bond shifts to 1730 cm−1, presumably caused by the conversion of CC to C–C as a result of polymerization, which suggests that NAQ is incorporated into the polyacrylate covalently. The peaks at 1665 cm−1 and 1594 cm−1 are assigned to skeleton vibrations of the aromatic rings whose intensities increase with the increasing ratio of NAQ. Other characteristic peaks assignment for PNAQ is as follows: 3015–2870 cm−1 (νCH3 and νCH2), 1485 cm−1 (δC–N) and 953 cm−1 (νC–N) of trimethylamino chloride, 1150 cm−1 (νC–O–C).
O bond conjugated to the CC bond. However, the stretching vibration peak of the CO bond shifts to 1730 cm−1, presumably caused by the conversion of CC to C–C as a result of polymerization, which suggests that NAQ is incorporated into the polyacrylate covalently. The peaks at 1665 cm−1 and 1594 cm−1 are assigned to skeleton vibrations of the aromatic rings whose intensities increase with the increasing ratio of NAQ. Other characteristic peaks assignment for PNAQ is as follows: 3015–2870 cm−1 (νCH3 and νCH2), 1485 cm−1 (δC–N) and 953 cm−1 (νC–N) of trimethylamino chloride, 1150 cm−1 (νC–O–C).
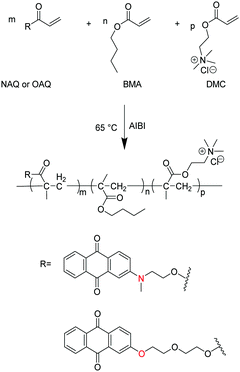 |
| | Scheme 1 Synthetic route toward single-component dual-emissive waterborne polyacrylates. | |
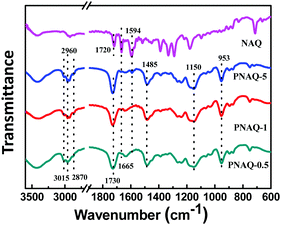 |
| | Fig. 1 FT-IR of PNAQ films with various NAQ weight percentage on a KBr plate (number denotes the weight percentage of NAQ, e.g. 0.5% NAQ = PNAQ-0.5; control represents polyacrylate without NAQ). | |
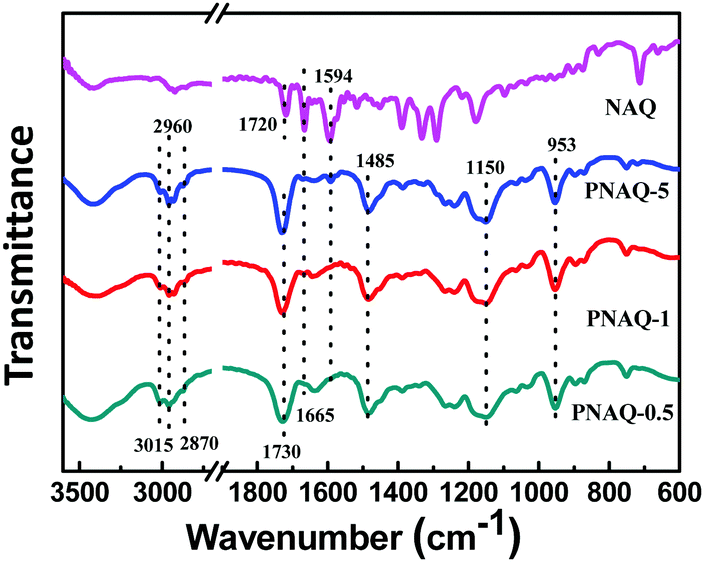
Shown in Fig. 2 are the thermal behaviors of PNAQ and POAQ polymers. From the differential scanning calorimetry (DSC, Fig. 2a and b) curves, all of the samples show a descending slope which is characteristic of the glass transition temperature (Tg). As the DSC curves show, Tg is around 115 °C for PNAQ and 118 °C for POAQ, which is similar to the control polymer (waterborne polyacrylate without NAQ or OAQ), suggesting that incorporation of NAQ or OAQ at a small percentage does not evidently change the motion of polymer side chains. From the thermogravimetric analysis (TGA, Fig. 2c and d), there are three apparent steps in the course of polyacrylate decomposition. All the polymer samples exhibit a slow descending trend from the onset to 240 °C due to the decomposition of the quaternary ammonium salt. A sharp descending shows between 240 °C and 300 °C, which indicates the breakage of the ester group. The polymeric main chains degrade at the final stage in between 300 °C and 450 °C. Therefore, NAQ and OAQ also do not obviously change the thermal behaviors of polyacrylates at up to 5% content.
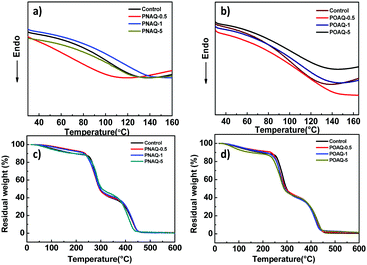 |
| | Fig. 2 DSC curves of polyacrylates with different NAQ (a) and OAQ (b) contents. TGA of polyacrylates with different NAQ (c) and OAQ (d) contents. | |
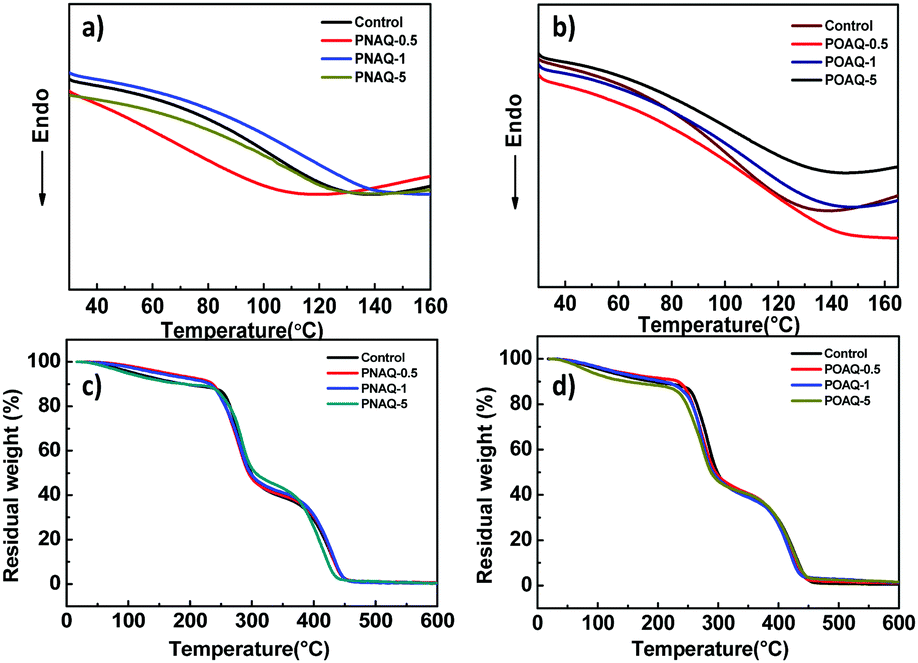
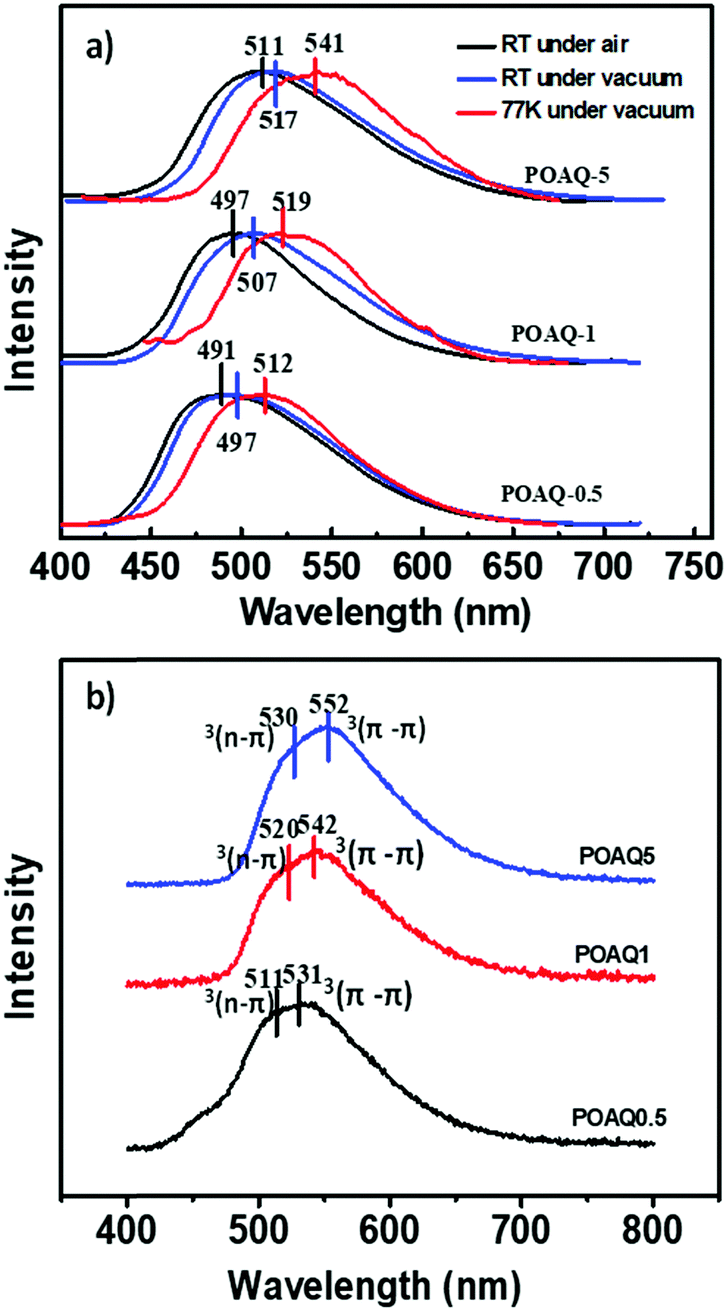
Since anthraquinone derivatives are generally weakly or non-luminescent in dilute solutions, the luminescence properties of the polymers were mainly investigated in the solid state. Compared to the steady-state emission spectrum of the POAQ-0.5 film in air (Fig. 3 and Table 1, λem = 491 nm) at room temperature, the maximal emission peak of the POAQ-0.5 film (λem = 497 nm) shifts bathochromically by ∼6 nm under vacuum, which is likely due to contribution from increased emissive triplet states at longer wavelengths. Correspondingly, the luminescence lifetime increases from 371.9 μs to 5.58 ms. Curiously, irrespective of the presence of oxygen, the measured lifetime is too long to be considered as typical fluorescence emission. We thus ascribe the emission from the POAQ-0.5 film to thermally activated delayed fluorescence (TADF). As a result, the steady-state emission is further red-shifted to 512 nm at 77 K, where the equilibrium favours more towards the radiative decay from a lower-energy state, i.e., the triplet state of OAQ. This is evidenced by a much longer-lived lifetime of 143.1 ms at this temperature at 512 nm. When the emission was collected after a 10 ms delay, an even more redshifted spectrum could be seen (λem = 531 nm). It is thus interesting to observe that two independently emitting triplet states (512 nm and 531, indicated by coloured lines) co-exist at 77 K.
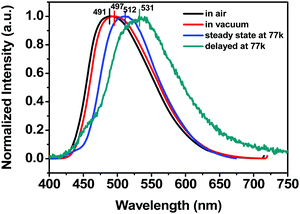 |
| | Fig. 3 Steady-state emission spectra (λex = 365 nm) of the POAQ-0.5 solid film under air at room temperature (black line), in vacuum at room temperature (red line) and 77 K (blue line) and the delayed emission spectrum at 77 K (Δt = 10 ms, cyan line). | |
Table 1 Luminescence data of POAQ films at room temperature in air and vacuum and at 77 K
|
|
λ
air
(nm) |
τ
air
(μs) |
λ
vc
(nm) |
τ
vc
(ms) |
λ
77K
(nm) |
τ
77K
(ms) |
τ
rad
(ms) |
Φ
(%) |
|
Steady-state emission maxima under air at room temperature.
Pre-exponent weight-averaged lifetimes under air.
Steady-state emission maxima under vacuum at room temperature.
Pre-exponent weight-averaged lifetimes under vacuum (triple exponential fitting).
Delayed emission maxima under vacuum at 77 K.
Pre-exponent weight-averaged lifetimes under vacuum at 77 K.
Calculated radiative lifetime under air.
Absolute emission quantum yields under air.
|
| POAQ-0.5 |
491 |
379.1 |
497 |
5.58 |
512 |
143.1 |
9.03 |
4.2 |
| POAQ-1 |
497 |
459.6 |
507 |
6.74 |
519 |
147.3 |
13.5 |
3.4 |
| POAQ-5 |
511 |
587.3 |
517 |
7.61 |
541 |
148.6 |
36.7 |
1.6 |
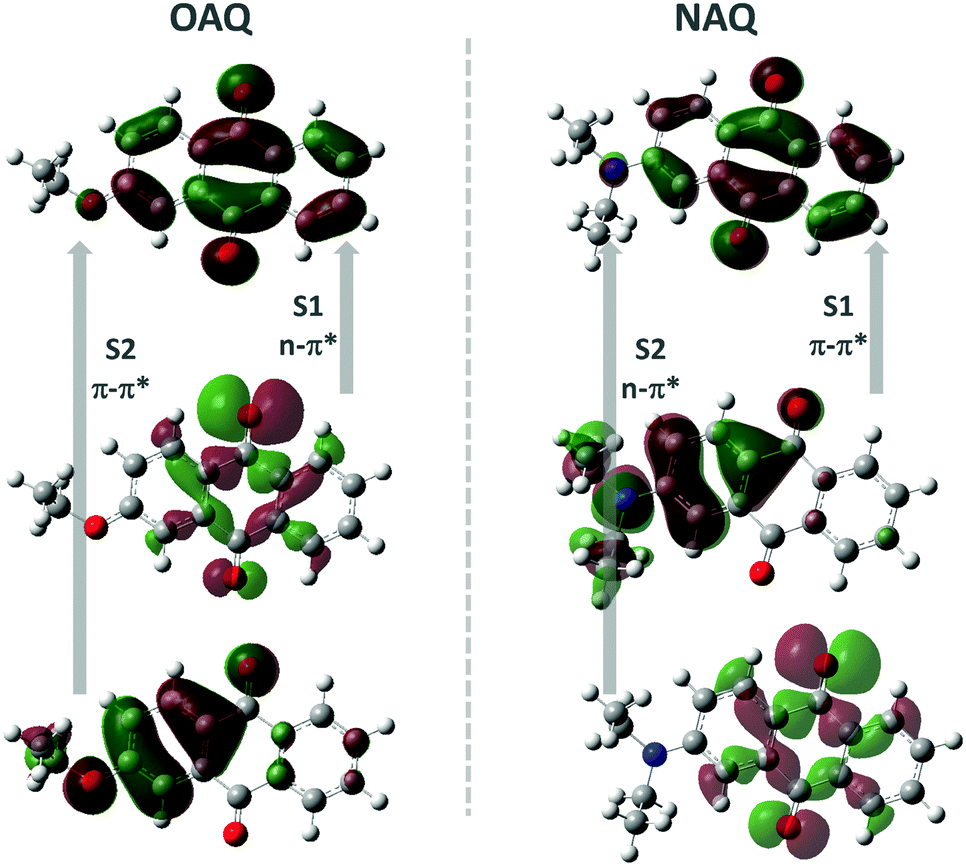
Quantum chemical calculations were performed to shed light on the possible emitting states of the AQ fluorophores (Fig. 4 and Table 2). The electronic emission spectrum (phosphorescence, triplet excited states transition to ground state, and fluorescence, singlet excited states transition to ground state) calculations are based on the Frank–Condon principle; the calculations are simulated by computing the energies of vertical transitions from excited states to the ground state with the equilibrium geometries of excited states. All the calculations were done using B3LYP/6-31+G* with the Gaussian 16 program package. From the geometry optimizations of excited states, the transition energies can be directly obtained (Table 2). The OAQ's second singlet excited state optimization cannot be obtained, while we can estimate the emission value to be larger than 425.86 nm (the second excited energy in the equilibrium geometry of the first excited states). From the calculations, the lowest singlet state (S1) has strong n–π* character while the second lowest one (S2) is π–π* with substantial charge-transfer (CT) character. Both n–π* and CT transitions favour the intersystem crossing (ISC) process to the triplet excited states, which may explain the dominant long-lived luminescence emission observed in the experiment.
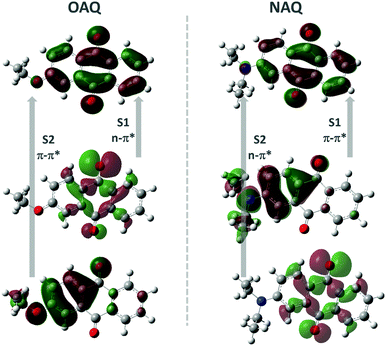 |
| | Fig. 4 Calculated molecular orbitals involved in the lowest two singlet and triplet states, where S1(2) denotes the first (second) singlet transition, respectively. | |
Table 2 The calculated excited energy values for OAQ and NAQ molecules using B3LYP/6-31+G*, where T1(2) denotes the first (second) triplet excited state and S1(2) denotes the first (second) singlet excited state
|
|
T1 |
T2 |
S1 |
S2 |
| OAQ |
642.16 nm |
586.84 nm |
475.07 nm |
>425.9 nm |
| 1.9307 eV |
2.1127 eV |
2.6098 eV |
<2.9114 eV |
| π–π* |
n–π* |
n–π* |
π–π* |
|
|
| NAQ |
788.94 nm |
593.73 nm |
601.75 nm |
484.13 nm |
| 1.5715 eV |
2.0882 eV |
2.0604 eV |
2.5610 eV |
| π–π* |
n–π* |
π–π* |
n–π* |
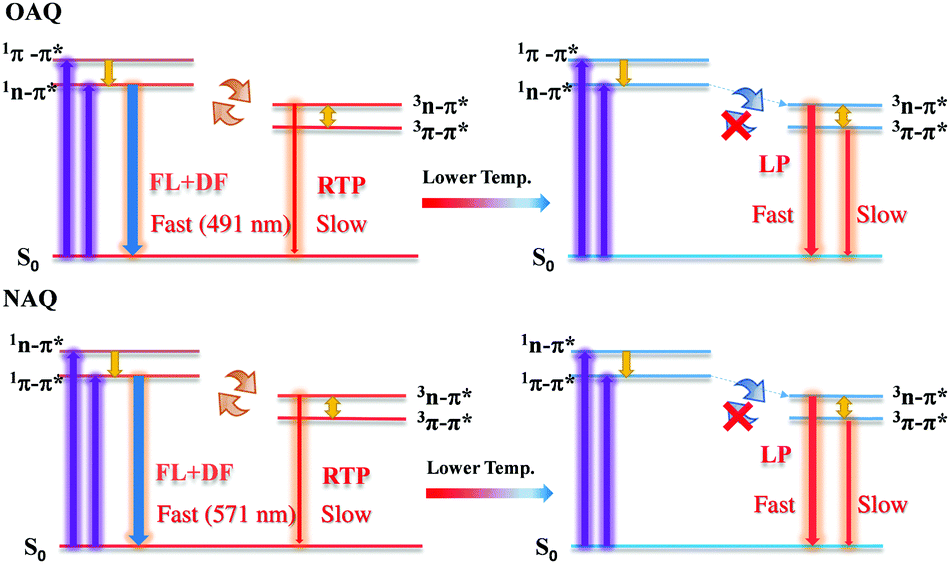
The proposed decay pathways for the excited states are shown in Scheme 2. The apparent lack of detectable fluorescence bands at room temperature indicates that the ISC process is incredibly efficient, due to the El-Sayed rule, and essentially quenches all singlet excited state populations. Based on the spectroscopic data in Fig. 3, the calculated energy gap between the lowest singlet state (S1, 491 nm) and the second triplet state (T2, 512), ΔES1T2, is 0.104 eV (or 2.39 kcal mol−1); the calculated energy gap between the lowest singlet state (S1, 491 nm) and the lowest triplet state (T1, 531), ΔES1T1, is 0.180 eV (or 4.15 kcal mol−1). And the calculated energy gap between the two triplet states, ΔET2T1, is 0.0867 eV (or 2.1 kcal mol−1). These values agree well with previous reports and allow for sufficient thermal population of all four states at room temperature. According to the equation r = exp(ΔE/T) (where r is the Boltzmann population, ΔE is the energy gap between the two emitting states and T is the temperature in Kelvin), the calculated Boltzmann population ratio at 298 K for ΔES1T2 is thus 0.0174:1 and 0.028:1 for ΔET2T1. The radiative decay rates for 1n–π*, 3n–π* and 3π–π*, however, are on the order of ns, μs, and ms, respectively. It is then not surprising to see that the majority of the radiative decay is from the TADF pathway. At 77 K, the Boltzmann ratios for the two scenarios are 1.65 × 10−7:1 (for ΔES1T2) and 1.10 × 10−6:1 (for ΔET2T1), respectively, where the upper emitting states can no longer compete with the lower one. Consequently, the dominant emission comes from the lower triplet states.
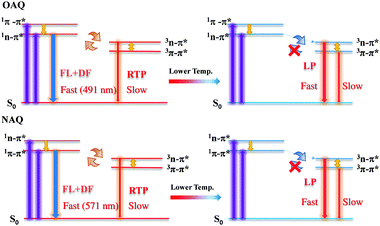 |
| | Scheme 2 Illustration of dominant emissive states at higher (left) and lower (right) temperatures with simplified Jablonski diagrams. | |
We then investigated the effects of dye concentration on luminescence properties, where three dye molar ratios (0.5%, 1% and 5%) were used for comparison (Fig. 5 and Table 1). From Fig. 5a, it is apparent that the emission maxima at both room temperature and 77 K are red-shifted as the OAQ content increases. The intrinsic luminescence lifetimes (τ/Φ, Table 1), however, show a dramatic increase from 9.03 ms (POAQ-0.5) to 13.5 ms (POAQ-1) and finally 36.7 ms for POAQ-5 at room temperature. The phenomenon can be explained by the splittings of excited state energy levels and generation of a relatively forbidden lowest emitting state from the dye molecule aggregates, as has been previous reported.28,29 From Fig. 4, the steady-state emission maxima change from 491 to 511 nm in air at room temperature; at low temperature, the change is more dramatic: from 512 to 541 nm. Apart from mechanisms similar to the previously reported β-diketone28 and benzophenone29 systems, in the POAQ system, the lengthened intrinsic lifetimes are also very likely due to increased mixing between the n–π* and π–π* states from dye aggregates.
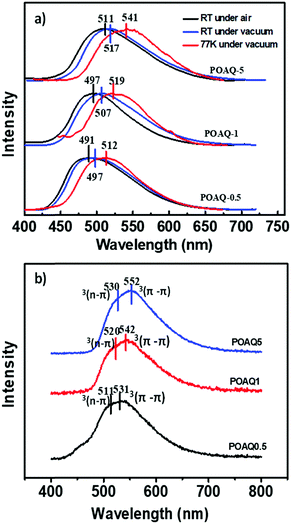 |
| | Fig. 5 (a) Steady-state emission spectra under air and vacuum of POAQ-0.5, POAQ-1, and POAQ-5 at room temperature and 77 K (λex = 365 nm). (b) Delayed emission spectra at 77 K under vacuum of POAQ-0.5, POAQ-1, and POAQ-5 (λex = 365 nm). | |
Next, the substitution effect was explored by replacing the alkoxyl group with alkylamino group on the AQ dye. Since the alkylamino group is a much stronger electron donor, two major effects can be expected from the nitrogen substitution: (1) the photoluminescence, both fluorescence and phosphorescence, should be red-shifted; (2) the luminescence emission quantum yields should be enhanced given the larger separation between 1n–π* and 1π–π*. From the calculations (Fig. 4 and Scheme 2), it is clear that the presence of the stronger donor N,N-substituted amino group has reversed the relative energy levels of the 1n–π* and 1π–π* states. Both effects can be easily observed from Fig. 5 and Table 3. From Table 3, the luminescence lifetime in air has been dramatically reduced to several microseconds despite a sharp increase in the measured quantum yield in the solid state. Compared to POAQ with dye concentrations at 0.5%, 1% and 5%, the emission maxima of PNAQ polymer films at these concentrations are red-shifted to 571, 593 and 611 nm, respectively, rendering the visual photoluminescence colour from green to orange-red (Fig. 6). Correspondingly, the phosphorescence emissions at 77 K were also substantially red-shifted. The quantum yields of the N-substituted AQ polymer films, PNAQs, also exhibit much larger values at 15.7%, 13.4% and 8.6% for PNAQ-0.5, PNAQ-1, and PNAQ-5, respectively. The calculated radiative lifetimes are thus reduced to a few tens of microseconds (35.9, 24.8 and 18.8 μs). We ascribe the phenomenon to the simultaneous increase in ISC and reverse ISC rates due to the closer energy levels between the lowest singlet state (1π–π*) and the second lowest triplet state (3n–π*), compared to the O-substituted dye. Curiously, the lifetimes measured in vacuum are significantly longer than the calculated intrinsic lifetimes. The anomaly is very likely due to the complete loss of the much-longer lived 3π–π* component in air, which contributes overwhelmingly (vs. lifetime for delayed fluorescence) in vacuum (Fig. 6).
Table 3 Luminescence data of PNAQ films in air and vacuum at room temperature and 77 K
|
|
λ
air
(nm) |
τ
air
(μs) |
λ
vc
(nm) |
τ
vc
(ms) |
λ
77K
(nm) |
τ
77K
(ms) |
τ
rad
(μs) |
Φ
(%) |
|
Steady-state emission maxima under air at room temperature.
Pre-exponent weight-averaged lifetimes under air.
Steady-state emission maxima under vacuum at room temperature.
Pre-exponent weight-averaged lifetimes under vacuum.
Delayed emission maxima under vacuum at 77 K.
Pre-exponent weight-averaged lifetimes under vacuum at 77 K.
Calculated radiative lifetime under air.
Absolute emission quantum yields under air.
|
| PNAQ-0.5 |
571 |
5.63 |
577 |
3.78 |
585 |
34.2 |
35.9 |
15.7 |
| PNAQ-1 |
593 |
3.32 |
605 |
2.95 |
609 |
60.2 |
24.8 |
13.4 |
| PNAQ-5 |
611 |
1.62 |
620 |
1.36 |
624 |
75.5 |
18.8 |
8.6 |
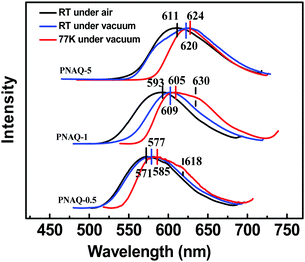 |
| | Fig. 6 Steady-state emission spectra under air and vacuum of PNAQ-0.5, PNAQ-1, and PNAQ-5 at room temperature and 77 K (λex = 365 nm). | |
Conclusions
In conclusion, we prepared a series of single-component dual-emissive waterborne polyacrylates containing O-substituted and N-substituted anthraquinone (PNAQs and POAQs). They both show thermally activated delayed fluorescence (TADF) which is found to be associated with the unique, two-fold triplet emissive states. The dye concentration is also shown to influence the luminescence properties of the polymers which can be explained with excited-state energy splittings.
Experimental
Materials
2-Chloroanthraquinone (98%, Aladdin Reagent Co.) was purified by recrystallization in ethanol before use. Butyl methacrylate (99%, BMA, Aladdin Reagent Co.) was eluted on alkaline aluminum oxide column chromatography to remove the polymerization inhibitor before use. Diethylene glycol (99%), N-methylaminoethanol (99%), 2-methacryloyl chloride (97%), 1-mercaptododecane (98%), 2,2′-azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride (98%, AIBI) and methacryloxyethyltrimethyl ammonium chloride (water solution, 80%, DMC) were purchased from Energy Chemical Reagent Co. and used without further purification. Other reagents were obtained from Sinopharm Chemical Reagent Co., Ltd and used as received.
Methods
1H NMR (300 MHz) and 13C NMR (75 MHz) spectra were recorded on a Bruker AV300 NMR spectrometer operated in the Fourier transform mode. NMR chemical shifts were reported in deuterated chloroform (CDCl3) with TMS as standard at room temperature. MALDI-TOF mass spectra (ESI) were obtained from an Atouflex Speed mass spectrometer. Fourier transform infrared (FT-IR) spectra were collected by using a Nicolet 6700 spectrophotometer and spectra were obtained from 4000 cm−1 to 400 cm−1. Differential scanning calorimetry (DSC) and thermogravimetric (TG) analysis spectra were investigated on Mettler-Toledo DSC and Shimadzu TGA-50 respectively. UV-Vis absorption spectra were recorded using UV-2401 manufactured by Shimadzu with 1 × 10−5 M dichloromethane. Excitation and steady-state fluorescence emission spectra and absolute quantum yield were recorded using a FluoroMax-4 spectrofluorometer (Horiba Scientific) and analyzed using the Origin integrated software FluoroEssence (v2.2). Fluorescence and phosphorescence lifetime data were acquired from a single photon counting controller with a 1 MHz LED laser with the excitation peak at 370 nm. Lifetime data were analyzed with DataStation v6.6 (Horiba Scientific) using triple-exponential fitting. Delayed photoluminescence spectra (77 K) were recorded on SpectraSuite (Ocean Optics, v2008) with 100 ms integrated time and 500 μs pulse signal under 365 nm excitation.
Synthesis
Synthesis of 2-[(2-hydroxyethyl)-methylamino]anthracene-9,10-dione.
To a 50 mL round-bottom flask equipped with a magnetic stir bar and a reflux condenser and containing 2.0 g of N-methylaminoethanol, 2.1 g of 2-chloroanthraquinone and 0.72 g of potassium hydroxide were added. Then the reaction was conducted at 130 °C for 10 hours. After the reaction finished, the mixture was cooled down to room temperature. Then the mixture was poured into 100 mL of deionized water in a 250 mL beaker and stirred for 1 h. The precipitate was collected by filtration in vacuum. The red brown crude product was subjected to recrystallization twice in ethanol and filtration for use in the next synthesis without further purification.
Synthesis of 2-methyl-acrylic acid 2-[(9,10-dioxo-9,10-dihydro-anthracen-2-yl)-methyl-amino]-ethyl ester (NAQ).
1.69 g of 2-[(2-hydroxyethyl)-methylamino]anthracene-9,10-dione, 0.91 g of triethylamine and 15 mL of dried dichloromethane were added to a 50 mL round-bottom flask with a magnetic stir bar. Then the mixture was stirred to dissolve all the solids at room temperature. 1.25 g of 2-methacryloyl chloride was dissolved in 5 mL of dichloromethane and added dropwise to the flask in the ice bath over 30 minutes. The reaction was performed at room temperature for 15 hours. After the reaction finished, the solution was extracted with water (50 mL) for three times. After drying over anhydrous Na2SO4, the solvent was removed in vacuum using a rotary evaporator. The crude product was further purified by column chromatography to obtain an orange-red product, yielding 1.91 g, 91%. 1H NMR (300 MHz, CDCl3) δ (TMS, ppm): 8.28 (td, 2H), 8.18 (d, 1H), 7.80–7.69 (m, 2H), 7.53 (d, 1H), 7.04 (dd, 1H), 6.12–6.00 (m, 1H), 5.55 (p, 1H), 4.41 (t, 2H), 3.86 (t, 2H), 3.20 (s, 3H), 1.89 (t, 3H). 13C NMR (75 MHz, CDCl3), δ (TMS, ppm): 184.07, 181.54, 167.18, 152.89, 135.74, 134.96, 134.25, 133.96, 133.65, 133.06, 129.82, 126.92, 126.87, 126.29, 122.44, 115.94, 108.52, 61.66, 50.68, 38.96, 18.28. MS (ESI): m/z [M + H]+ calcd for C21H20O4N 350.13868, found 350.13831.
Synthesis of 2-[2-(2-hydroxyethoxy)ethoxy]anthracene-9,10-dione.
This synthetic process is similar to the synthesis of 2-[(2-hydroxyethyl)-methylamino]anthracene-9,10-dione. The crude product was subjected to recrystallization twice in ethanol and filtration for use in the next synthesis without further purification.
Synthesis of 2-{2-[(9,10-dioxo-9,10-dihydroanthracen-2-yl)oxy]ethoxy}ethyl methacrylate (OAQ).
This synthetic process is similar to the synthesis of 2-methyl-acrylic acid 2-[(9,10-dioxo-9,10-dihydro-anthracen-2-yl)-methyl-amino]-ethyl ester (NAQ), yielding 3.08 g, 81.2%. 1H NMR (300 MHz, CDCl3), δ (TMS, ppm): 8.35–8.22 (m, 3H), 7.86–7.71 (m, 3H), 7.31 (dd, 1H), 6.17–6.09 (m, 1H), 5.62–5.53 (m, 1H), 4.34 (td, 4H), 3.90 (dt, 4H), 1.56 (s, 3H). 13C NMR (75 MHz, CDCl3), δ (TMS, ppm): 183.18, 182.12, 167.34, 163.49, 136.07, 135.53, 134.17, 133.68, 133.64, 133.55, 129.75, 127.14, 125.87, 121.6, 110.54, 77.22, 69.45, 69.35, 68.10, 63.72, 18.32. MS (ESI): m/z [M + H]+ calcd for C22H21O6 381.13326, found 381.13284.
Polymerization of NAQ (PNAQ).
60 mg of NAQ, 2.0 g of BMA and 2.5 g of acetone were added into a three-neck round-bottom flask equipped with a mechanical stir bar and a thermometer. Then the reaction was heated to 65 °C and stirred until NAQ was dissolved. 12.5 g of DMC solution, 0.48 g of NDM and 12.5 g of deionized water were added to the flask. Then the reaction was allowed to run at 65 °C and at a stirring rate of 300 rpm. 120 mg of AIBI was dissolved in water and added to the flask dropwise over 2 hours. After the complete addition of AIBI, the reaction was continued to proceed at 65 °C for 7 hours. After the reaction finished, acetone was removed in vacuum using a rotary evaporator. Finally, orange-red DMC, BMA and NAQ copolymer emulsion containing 0.5 wt% NAQ was obtained (PNAQ-0.5). PNAQs with other weight percentages of NAQ were prepared similarly. The specific ratio of each component is listed in Table S1 (ESI†). The formation of PNAQ films was on the polytetrafluoroethylene panel until water evaporated.
Polymerization of OAQ (POAQ).
60 mg of OAQ, 2.0 g of BMA and 2.5 g of acetone were added into a three-neck round-bottom flask equipped with a mechanical stir bar and a thermometer. Then the reaction was heated to 65 °C and stirred until OAQ was dissolved. 12.5 g of DMC solution, 0.48 g of NDM and 12.5 g of deionized water were added to the flask. Then the reaction was continued to proceed at 65 °C and at a stirring rate at 300 rpm. 120 mg of AIBI was dissolved in water and added to the flask dropwise over 2 hours. After the complete addition of AIBI, the reaction was continued to proceed at 65 °C for 7 hours. After the reaction finished, acetone was removed in vacuum using a rotary evaporator. Finally, orange-red DMC, BMA and OAQ copolymer emulsion containing 0.5 wt% OAQ was obtained (POAQ-0.5). POAQs with other weight percentages of OAQ were prepared similarly. The specific ratio of each component is listed in Table S2 (ESI†). The formation of POAQ films was on the polytetrafluoroethylene panel until water evaporated.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
We acknowledge support from the Fundamental Research Funds for the Central Universities (WK2340000068 to G. Z.) and the National High Technology Research and Development Program of China (2015AA033903 to X. Z.).
Notes and references
- F. Yu, X. Han and L. Chen, Chem. Commun., 2014, 50, 12234–12249 RSC.
- X. Xiong, L. Zheng, J. Yan, F. Ye, Y. Qian and F. Song, RSC Adv., 2015, 5, 53660–53664 RSC.
- X. Chen, C. Xu, T. Wang, C. Zhou, J. Du, Z. Wang, H. Xu, T. Xie, G. Bi, J. Jiang, X. Zhang, J. N. Demas, C. O. Trindle, Y. Luo and G. Zhang, Angew. Chem., Int. Ed., 2016, 55, 9872–9876 CrossRef CAS PubMed.
- D. Lee, J. Jung, D. Bilby, M. S. Kwon, J. Yun and J. Kim, ACS Appl. Mater. Interfaces, 2015, 7, 2993–2997 CAS.
- Z. Yu, Y. Wu, L. Xiao, J. Chen, Q. Liao, J. Yao and H. Fu, J. Am. Chem. Soc., 2017, 18, 6376–6381 CrossRef PubMed.
- Y. Seino, H. Sasabe, Y. J. Pu and J. Kido, Adv. Mater., 2014, 26, 1612–1616 CrossRef CAS PubMed.
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS.
- O. Bolton, K. Lee, H.-J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205–210 CrossRef CAS PubMed.
- D. Lee, O. Bolton, B. C. Kim, J. H. Youk, S. Takayama and J. Kim, J. Am. Chem. Soc., 2013, 135, 6325–6329 CrossRef CAS PubMed.
- G. Zhang, J. Chen, S. J. Payne, S. E. Kooi, J. Demas and C. L. Fraser, J. Am. Chem. Soc., 2007, 129, 8942–8943 CrossRef CAS PubMed.
- G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747–751 CrossRef CAS PubMed.
- S. Kim, H. J. Kwon, S. Lee, H. Shim, Y. Chun, W. Choi, J. Kwack, D. Han, M. Song and S. Kim, Adv. Mater., 2011, 23, 3511–3516 CrossRef CAS PubMed.
- Z. Yu, X. Niu, Z. Liu and Q. Pei, Adv. Mater., 2011, 23, 3989–3994 CrossRef CAS PubMed.
- M. Pope, H. Kallmann and P. Magnante, J. Chem. Phys., 1963, 38, 2042–2043 CrossRef CAS.
- S. Hirata, Y. Sakai, K. Masui, H. Tanaka, S. Y. Lee, H. Nomura, N. Nakamura, M. Yasumatsu, H. Nakanotani, Q. Zhang, K. Shizu, H. Miyazaki and C. Adachi, Nat. Mater., 2015, 14, 330–336 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- X. K. Liu, Z. Chen, J. Qing, W. J. Zhang, B. Wu, H. L. Tam, F. Zhu, X. H. Zhang and C. S. Lee, Adv. Mater., 2015, 27, 7079–7085 CrossRef CAS PubMed.
- J. Luo, S. Gong, Y. Gu, T. Chen, Y. Li, C. Zhong, G. Xie and C. Yang, J. Mater. Chem. C, 2016, 4, 2442–2446 RSC.
- S. Chen, Y. Hong, Y. Liu, J. Liu, C. W. Leung, M. Li, R. T. Kwok, E. Zhao, J. W. Lam, Y. Yu and B. Z. Tang, J. Am. Chem. Soc., 2013, 135, 4926–4929 CrossRef CAS PubMed.
- J. Luo, Z. Xie, J. W. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- R. S. Singh, R. K. Gupta, R. P. Paitandi, M. Dubey, G. Sharma, B. Koch and D. S. Pandey, Chem. Commun., 2015, 51, 9125–9128 RSC.
- H. Li, X. Zhang, Z. Chi, B. Xu, W. Zhou, S. Liu, Y. Zhang and J. Xu, Org. Lett., 2011, 13, 556–559 CrossRef CAS PubMed.
- Z. Ren, R. S. Nobuyasu, F. B. Dias, A. P. Monkman, S. Yan and M. R. Bryce, Macromolecules, 2016, 49, 5452–5460 CrossRef CAS.
- A. E. Nikolaenko, M. Cass, F. Bourcet, D. Mohamad and M. Roberts, Adv. Mater., 2015, 27, 7236–7240 CrossRef CAS PubMed.
- Y. Zhu, Y. Zhang, B. Yao, Y. Wang, Z. Zhang, H. Zhan, B. Zhang, Z. Xie, Y. Wang and Y. Cheng, Macromolecules, 2016, 49, 4373–4377 CrossRef CAS.
- S. L. Chai and M. M. Jin, J. Appl. Polym. Sci., 2009, 114, 2030–2035 CrossRef CAS.
- X. Zhou, W. Tu and J. Hu, Chin. J. Chem. Eng., 2006, 14, 99–104 CrossRef CAS.
- X. Sun, X. Wang, X. Li, J. Ge, Q. Zhang, J. Jiang and G. Zhang, Macromol. Rapid Commun., 2015, 36, 298–303 CrossRef CAS PubMed.
- C. Zhou, T. Xie, R. Zhou, C. O. Trindle, Y. Tikman, X. Zhang and G. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 17209–17216 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: FT-IR, 1H and 13NMR. See DOI: 10.1039/c7qm00399d |
|
| This journal is © the Partner Organisations 2018 |

 ab,
Zhen
Tang
c,
Dong
Xu
a,
Wei
Sun
a,
Yipeng
Deng
a,
Qidong
Wang
a,
Xingyuan
Zhang
*a,
Peifeng
Su
ab,
Zhen
Tang
c,
Dong
Xu
a,
Wei
Sun
a,
Yipeng
Deng
a,
Qidong
Wang
a,
Xingyuan
Zhang
*a,
Peifeng
Su