
A fast route to obtain modified tin oxide aerogels using hydroxostannate precursors†
Max Gregor
Beier
a,
Christoph
Ziegler
a,
Karl
Wegner
b,
Albrecht
Benad
a,
Frank
Simon
 c,
Stefan
Kaskel
b and
Alexander
Eychmüller
*a
c,
Stefan
Kaskel
b and
Alexander
Eychmüller
*a
aPhysical Chemistry, TU Dresden, Bergstraßee 66b, 01062 Dresden, Germany. E-mail: alexander.eychmueller@chemie.tu-dresden.de
bInorganic Chemistry, TU Dresden, Bergstraße 66, 01062 Dresden, Germany
cLeibnitz Institute for Polymer Research Dresden, Hohe Straße 6, 01069 Dresden, Germany
First published on 29th January 2018
Abstract
Nanostructured tin oxide materials with a high specific surface area and porosity are promising for applications such as electrocatalysis, lithium ion batteries or sensors. Here, we present a facile strategy for the synthesis of tin oxide aerogels using inexpensive hexahydroxostannate as a tin precursor. This easy and scalable method yields tin oxide aerogels with a high specific surface area and a wide pore size distribution. The method can be modified by adding hexahydroxoantimonate to obtain antimony-doped tin oxide aerogels that exhibit electrical conductivity after annealing. Cogelation with other preformed nanoparticles (e.g. Au, Pt) leads to mixed gels. Both modifications do not have a large impact on the porous properties of the obtained aerogels. Tin oxide materials prepared via this route can be tailored to a specific application by versatile modification possibilities.
Introduction
Tin(IV) oxide is a wide-bandgap semiconductor and has been investigated for many applications such as anode materials for Li ion batteries,1,2 gas sensors,3 dye sensitized solar cells4 and different fields of catalysis, photocatalysis5,6 and electrocatalysis.7–9 To obtain good performance in these applications it is crucial to provide a large specific surface area to enhance exchange reactions. For this purpose, nanostructured materials are used. A pore system is important to enable diffusion of Li ions, analyte molecules or reactants through the material. Aerogels combine both aspects; they have a high surface area and a wide pore size distribution that allows fast transport.Tin oxide can be modified in specific ways to increase its versatility for certain applications. For example, it can be doped with antimony to obtain electrically conductive antimony-doped tin oxide (ATO) for electronic or electrocatalytic applications. Metal nanoparticles can be loaded onto the material to optimize its sensitivity, response time and selectivity of the sensor materials or provide active centers in catalysts.
There are several approaches reported to synthesize tin oxide aerogels. Wu et al. described an all inorganic route, where spontaneous formation of a tin oxide gel is induced by increasing the pH value which was done by diluting a solution of tin(IV) chloride in a water/ethanol mixture.10 Most other published methods require organic reactants. Like many other metal oxides, tin oxide gels can be formed by hydrolysis of tin alkoxides.11 By adding antimony alkoxide to this hydrolyzation route, conductive antimony-doped tin oxide aerogels can be obtained.8 Furthermore, the ATO gels can be loaded with palladium particles with a post preparative treatment to obtain a potential fuel cell catalyst.12 Harreld et al. synthesized aerogels via a non-hydrolytic approach by the condensation of tin alkoxides and tin chlorides.13 Baumann et al. presented a facile method to synthesize tin oxide aerogels from inexpensive tin(IV) chloride, but they used carcinogenic and highly volatile propene oxide.14 This method was modified by Correa-Baena by adding an antimony precursor to obtain conductive ATO aerogels.15,16 The route by Rechberger et al. to conductive ATO aerogels includes several steps.17 First, they prepared nanoparticles in organic solvents assisted by microwave radiation. A highly concentrated aqueous suspension of these particles can be gelled by a heat treatment.
However, to date there is no report on a route leading to modified tin oxide aerogels that do not require expensive or dangerous reactants and that can be performed with less effort. Using inorganic reactants and water as the solvent is desirable, because of low cost and less impact on the environment. Tin chlorides are inexpensive and soluble precursors for an aqueous approach, but adsorbed halides from the precursor could have a negative influence on the sintering behavior of metal oxides during thermal treatment.18–20
In this study we present a very facile, all-inorganic route to high surface area tin oxide aerogels. The tin precursors used in this halide-free method are sodium or potassium salts of the hexahydroxostannate(IV) anion. It is known that solutions of hexahydroxostannate(IV) form gel like precipitates if two equivalents of acid per equivalent of tin are added.21 But so far, these hydrogels have not been supercritically dried to obtain tin oxide aerogels.
Results and discussion
Formation of tin oxide sol
Adding a weak coordinating acid such as nitric or acetic acid to an aqueous solution of hexahydroxostannate results in a turbid solution. When less than 2 equivalents (eq.) are added, the solution becomes clear again after some minutes. If more than 2 eq. of acid are added, the turbidity does not vanish and a hydrogel sediments. The turbidity indicates the formation of particles and large aggregates. The vanishing of the turbidity in the case of less than 2 eq. of H+ added indicates the dissolution of these aggregates. The resulting particle sol is stable for weeks even at a high concentration of up to 1 wt%.The dynamic light scattering (DLS) of the sol measured at least one hour after the addition of nitric acid shows small particles with an average diameter of 3.2 nm, and the distribution is broad with a standard deviation (σ) of 1 nm (Fig. 1).
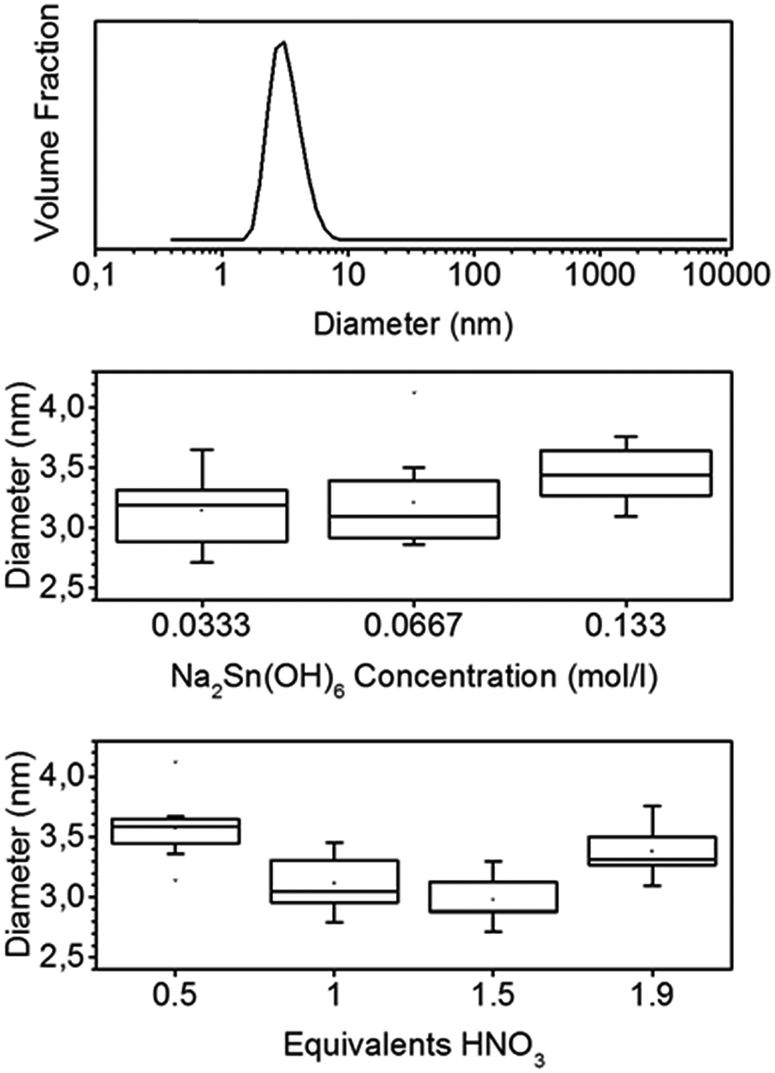 | ||
| Fig. 1 Representative particle size distribution of tin oxide sol measured by DLS (top) and boxplots of mean particle diameters at different hydroxostannate concentrations (center) and number of HNO3 equivalents (bottom). | ||
Moreover, the dependence of the particle size on the stannate concentration and equivalents of nitric acid was investigated. The mean diameters, determined by DLS, are shown in the boxplots of Fig. 1. The mean diameters range from 2.8 to 4.1 nm. The σ values of the individual size distributions vary from 0.8 to 1.2 nm. A high stannate concentration of 0.133 mol L−1 (2 wt% if fully transformed to SnO2) leads to a sol with larger particles. The minimum average particle size of 3 nm is observed for a number of 1.5 eq. of nitric acid. A low amount of 0.5 eq. or a large amount of 1.9 eq. of nitric acid, which is close to the point where gelling starts, leads to larger particles of 3.6 and 3.3 nm, respectively.
Gel formation
The sol used for further studies is obtained by the addition of 1.5 eq. of nitric acid. It can be destabilized to form a voluminous and turbid hydrogel by the second addition of acid to a total of more than 2 eq. of H+ per stannate ion. The duration of the gelation by the second addition of acid depends on the type and amount of acid and can further be controlled by temperature. As can be seen in Table 1, gel formation always occurs almost immediately if the pH during gelation is in the range from 2 to 3 either by the addition of weak acetic acid or low excess of nitric acid. If a large excess amount of strong nitric acid is added, resulting in pH value below 1, the duration of gel formation is expanded to several minutes. Only in this case the duration can be significantly influenced by temperature.| Acid | Total number of H+ eq. | Resulting pH | Temperature | Gelation timea |
|---|---|---|---|---|
| a Approximated duration until maximum turbidity and the start of flocculation or sedimentation of the hydrogel. | ||||
| Nitric acid HNO3 | 2.5 | 2.3 | RT | 10 s |
| 5 | 0.7 | RT | 30–45 min | |
| 0 °C | 4–6 h | |||
| 70 °C | 30 s | |||
| Acetic acid HOAc | 2.5 | 3 | RT | <5 s |
| 5 | 2.8 | RT | <5 s | |
| 0 °C | 10 s | |||
| 70 °C | <5 s | |||
The dependence of gelation time on pH can be explained by the colloidal stability of the charge-stabilized tin oxide particles. Zeta potential measurements on ATO particle dispersions by Li et al. show that the zeta potential and colloidal stability are minimal in the pH range from 2 to 3 and increase for pH values lower than 2.22 Besides, the particle sol can be destabilized to form a gel by the addition of organic solvents like acetone or ethanol. In this case the gel formation takes place within seconds. In contrast to the hydrogels formed by acid addition, these solvogels are compact and opaquely white.
For the ease of preparation, the gel formation with an excess of nitric acid at room temperature is preferred. The gelation time is relatively short but allows complete mixing without damaging the forming gel structure by stirring.
The presented route is a fast and convenient method to obtain tin oxide aerogels. Hexahydroxostannates are inexpensive tin precursors and only water as a solvent and diluted acids as low hazardous reagents are needed. For supercritical drying (SCD) the exchange with acetone and carbon dioxide is necessary. The method can easily yield material in large scale, just limited by the supercritical drying process.
Characterization of pure tin oxide gels
Fig. 2 shows the powder X-ray diffractograms (XRD) of gels formed by the addition of nitric acid and acetone in comparison. Both patterns exhibit only reflexes of cassiterite type SnO2. This shows that tin(IV) oxide particles are formed after the first addition of acid and that these do not change if gelation is induced by the second addition of acid. The very strong reflex broadening indicates very small particles or poor crystallinity.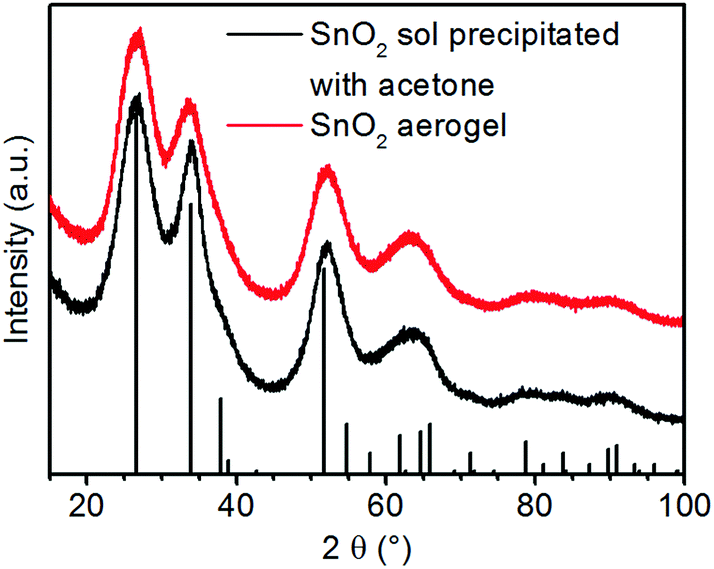 | ||
| Fig. 2 X-ray diffractograms of the precipitated tin oxide sol and tin oxide aerogel after SCD (vertical black bars: the reference pattern of the tetragonal cassiterite structure of SnO2). | ||
The nitrogen physisorption isotherm for the obtained aerogels is typical for aerogels (Fig. 3, top). According to the IUPAC classification, it is a type II isotherm with multilayer adsorption and no plateau at high relative pressures, which is a characteristic of samples containing macropores.23 The hysteresis indicates capillary condensation in the mesopores. The specific surface area (SSA) of aerogels prepared by the addition of acid at room temperature is in the range of 340–375 m2 g−1, which is close to the best reported values of 383 m2 g−1 for the tin oxide aerogels obtained via the epoxide method.14 The SSA of the aerogels prepared by the addition of acid at 0 °C or 70 °C is lower and ranges from 267 to 330 m2 g−1. Gels obtained by the addition of ethanol to a tin oxide sol have a much lower SSA of 110 m2 g−1. The tin oxide aerogels display a typical wide pore size distribution, with the maximum around 10 nm (Fig. 3, bottom).
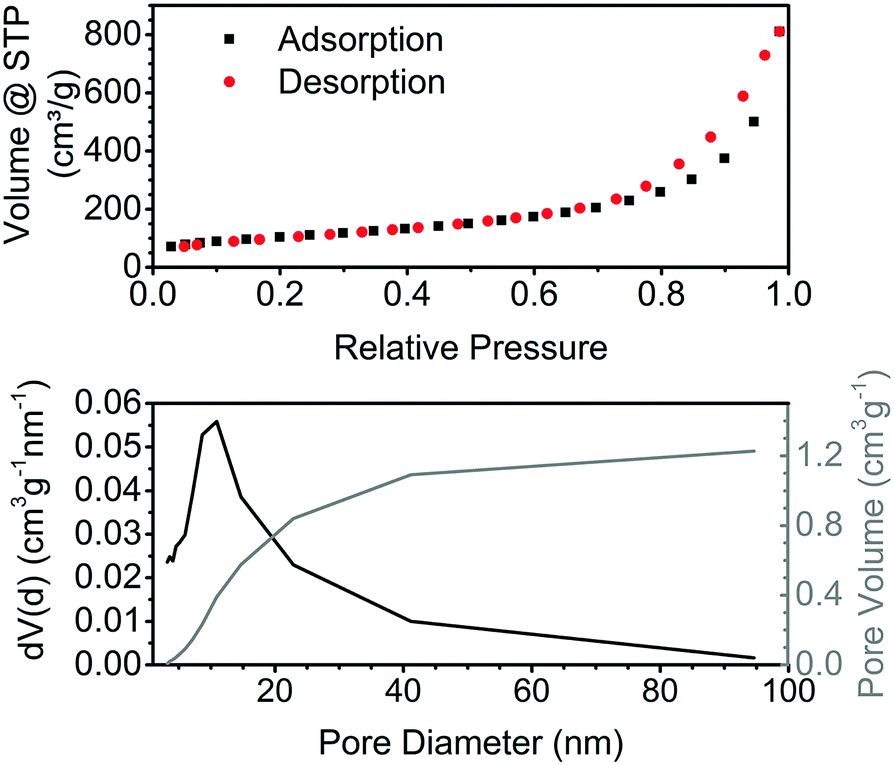 | ||
| Fig. 3 Nitrogen physisorption isotherm at 77 K of pure tin oxide aerogel and pore size distribution calculated using the BJH theory. | ||
Scanning and transmission electron microscopy (SEM and TEM) was used to characterize the morphology of the gels. SEM investigations reveal a sponge-like structure with a large variety of pore sizes (Fig. 4a). The TEM images in Fig. 4b and c show that the gel formed by the addition of acid is a fine network of small primary particles of 3–4 nm in diameter. The diameter of the primary particles in the gel matches very well with the nanoparticle diameter obtained from DLS measurements of the initial sol. Fig. 4d shows the structure of an aerogel formed via the destabilization of a tin oxide sol by applying ethanol. The primary particles form large aggregates that are connected to a network. The compact aggregates exhibit a small internal surface area, confirmed by nitrogen physisorption measurements.
 | ||
| Fig. 4 Electron microscopy images showing the morphology of the aerogels. (a) SEM micrograph of an aerogel obtained by gelation with nitric acid. (b and c) TEM micrographs of aerogels obtained by gelation with nitric acid. (d) TEM micrograph of an aerogel obtained by gelation with ethanol. | ||
Conductive ATO aerogels
The stannate route to obtain tin oxide can easily be modified by adding hexahydroxoantimonate(V) to the hexahydroxostannate solution prior to the first addition of acid. The duration of the gelation upon the second addition of acid is shortened with increasing amount of antimony to ten seconds at room temperature when the Sb content is 10 at%. Please note that the addition of acid to a solution of hexahydroxoantimonate without hexahydroxostannate leads to the formation of a compact precipitate but not a hydrogel.After supercritical drying the resulting ATO aerogels appear completely white. Upon subsequent annealing at 550 °C in air, the ATO gels turn blue. This indicates the incorporation of Sb(V) ions in the tin oxide lattice, which leads to the formation of oxygen vacancies. The resulting degenerated n-type semiconductor material has a plasmon absorption onset in the orange to red part of the visible spectrum.24 Hence, the blue color is more intense for samples containing more Sb.
The atomic ratio of Sb to Sn was studied by energy dispersive X-ray (EDX) analysis for the samples with precursor ratios of 5 and 10%, respectively. The result of the analysis was a Sb/Sn ratio of 4.5 ± 0.4 at% and 10.1 ± 0.6 at%, respectively. Accordingly, the Sb from the hexahydroxoantimonate precursor is completely incorporated in the resulting gels.
The conductivity of the calcined ATO gels was characterized in a pressure cell similar to the one deployed by Ozouf and Beauger.8 The measured specific conductivities are listed in Table 2.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
The SSA of the ATO gels after supercritical drying and after calcination is shown in Fig. 5. The SSA of the tin oxide aerogels after supercritical drying is only slightly decreased by the addition of antimony, but it is still larger than 300 m2 g−1. After calcination at 550 °C the aerogels retain a SSA of approximately 100 m2 g−1. Furthermore, samples with a higher antimony content exhibit larger SSAs.
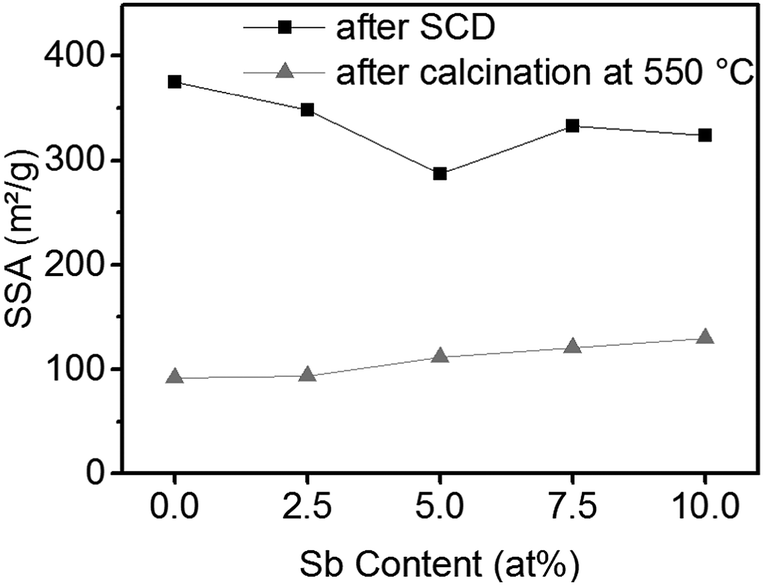 | ||
| Fig. 5 SSA of ATO aerogels after SCD and after calcination at 550 °C. The SSA of the non-calcined aerogels decreases from 375 m2 g−1 to 324 m2 g−1 with increasing Sb content. In contrast, the SSA of the calcined aerogels increases from 92 to 130 m2 g−1 with increasing Sb content. | ||
Table 3 shows the specific conductivity and SSA of the ATO aerogel with 10 at% Sb obtained via the stannate route in comparison to the ATO gels reported in the literature. The ATO gel obtained by the stannate route reaches the same conductivity as samples prepared by Ozouf and Beauger or Rechberger et al., which needed harsher calcination temperatures and a UV radiation treatment for 30 h prior to calcination in the latter to obtain good conductivity, respectively. Rechberger et al. reported on ATO aerogels where actually the samples with 5 at% Sb showed better conductivity, but the UV treatment was only done with samples containing 10 at% Sb. The ATO aerogels from the stannate route exhibited the largest SSA, due to mild temperatures and a large SSA before calcination.
| Reference | Sb content (precursors) (mol%) | Treatment/calcination | Specific conductivity (S cm−1) | SSA (m2 g−1) |
|---|---|---|---|---|
| Stannate route | 10 | 550 °C/2 h | 0.14 | 130 |
| Ozouf and Beauger8 | 10 | 600 °C/6 h | 0.15 | 87 |
| Rechberger et al.17 | 10 | UV + 500 °C/2 h | 0.027 | 117 |
| UV + 650 °C/2 h | 0.15 | 90 | ||
| Correa-Baena et al.16 | 20 | 800 °C/6 h | 0.071 | 38 |
| 30 |
The XRD patterns of the calcined ATO samples as shown in Fig. 6 only show reflexes of cassiterite SnO2. In contrast to the calcined ATO aerogels prepared by Ozouf and Beauger, no significant shift for the positions of the reflections is observed.8 Owing to the thermal treatment, the crystallites fuse to larger particles, which results in narrower reflexes than those observed for the aerogel right after supercritical drying. The FWHM of the reflections increases with the Sb doping level. For the (1 1 0)-reflection it changes from 1.60° for the calcined pure tin oxide gel to 2.07° for the sample containing 10 at% Sb. This indicates smaller crystallite size of samples that contain more antimony, which explains the higher SSA.
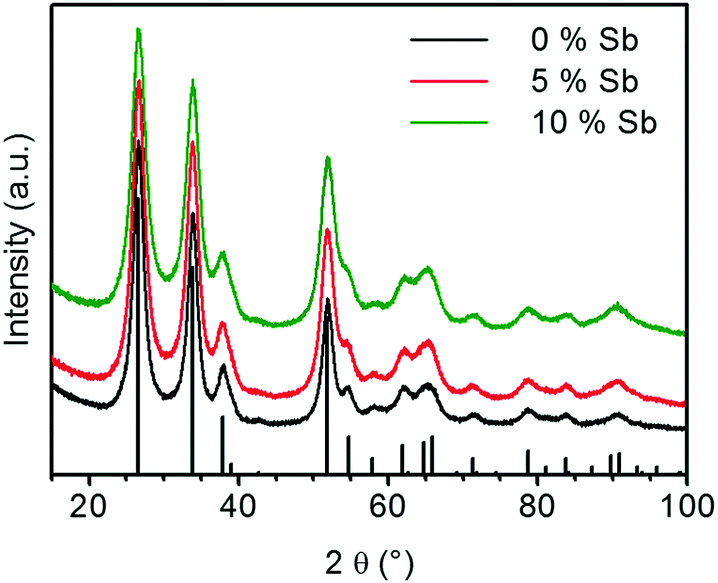 | ||
| Fig. 6 X-ray diffractograms of calcined ATO aerogels (vertical black bars: the reference pattern of the tetragonal cassiterite structure of SnO2). | ||
The effects of the antimony content on crystallite size and SSA are in agreement with the results of Rechberger et al. and Correa-Baena et al., who also found that the crystallite size is smaller and the retained SSA of the calcined aerogels is larger for samples with a higher antimony content.16,17
Incorporation of noble metal particles by cogelation
Citrate stabilized noble metal particles were synthesized via aqueous routes using sodium borohydride as a strong reducing agent.25,26 These yield small spherical gold particles with a diameter of 4–5 nm and platinum particles with a diameter of 3–4 nm, respectively. The sols of the metal particles can be directly mixed with the tin oxide particle sol without any treatment like washing, exchange or removal of the stabilizer, keeping this method very simple. When a noble metal nanoparticle solution is mixed with a stannate solution, the colloidal stability is decreased due to the high pH value and ionic strength. Nevertheless the noble metal particles are not destabilized upon mixing with the tin oxide sol as the ionic strength and pH value of the stannate solution decreased to moderate values by the acid addition to form the tin oxide particles.The second addition of acid destabilizes tin oxide and noble metal particles simultaneously leading to the gel formation. As shown in the TEM image in Fig. 7, the excess of tin oxide particles prevents the formation of aggregates of metal particles leading to a homogenous dispersion of noble metal particles on the tin oxide gel, even with a noble metal loading of 10 wt%.
 | ||
| Fig. 7 TEM brightfield micrographs of mixed aerogels containing 10 wt% Pt particles (left) and 5 wt% Au particles (right). Due to a higher density and higher atomic number the noble metal particles appear darker. | ||
The noble metal contents were determined for Au 5.4 ± 0.6 wt% and for Pt 5.6 ± 0.3 wt% and 10.9 ± 1.1 wt% Pt, respectively, by EDX analysis. This matches the targeted compositions of 5 wt% and 10 wt%, respectively.
The SSA of the mixed aerogel only slightly decreased upon the addition of sodium citrate or citrate stabilized noble metal particles, as shown in Table 4.
| Sample | Pure SnO2 | SnO2 + citrate | 5 wt% Au | 5 wt% Pt | 10 wt% Pt |
|---|---|---|---|---|---|
| SSA (m2 g−1) | 342 | 310 | 335 | 320 | 302 |
The XRD patterns of the aerogel loaded with platinum particles after SCD in as shown in Fig. 8 show additional reflections at 39.8° and 46.2° that correspond to the (1 1 1) and (2 0 0) lattice planes of platinum, respectively. The reflexes are more pronounced for the sample with a higher loading of 10 wt%. The peaks are broadened due to the small size of platinum particles (similar for Au/SnO2, not shown here).
 | ||
| Fig. 8 X-ray diffractogram of 10% Pt/SnO2–aerogel after SCD (vertical black and red bars: the reference patterns of tetragonal cassiterite structures of SnO2 and fcc Pt, respectively). | ||
To determine the oxidation state of the elements in the mixed noble metal/tin oxide aerogels, XPS was performed on pure SnO2 and Pt/SnO2 and Au/SnO2 aerogels. The recorded spectra are shown in Fig. 9. The binding energy values found for the Sn 3d peaks corresponded with 487.0 eV (Sn 3d5/2 peak) and 495.4 eV (Sn 3d3/2 peak) to the values expected for tin oxides. XPS seemed to be unsuitable to distinguish between tin(II) and (IV) in oxides.
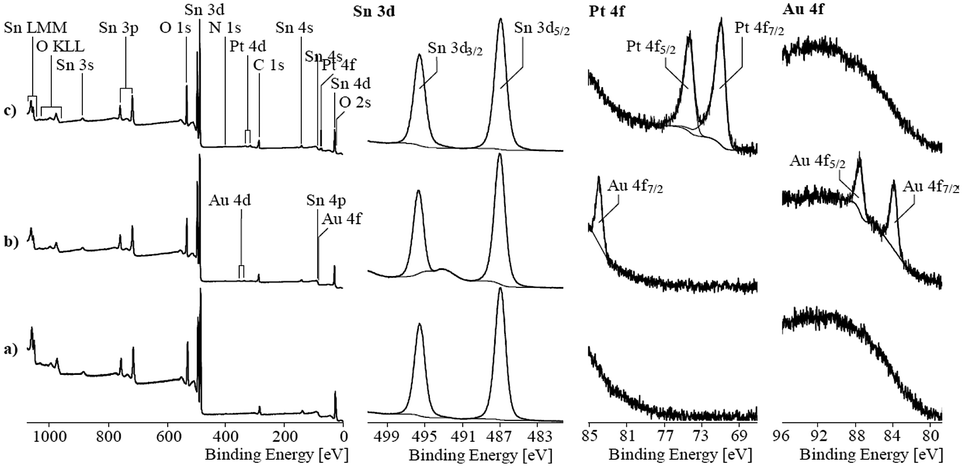 | ||
| Fig. 9 X-ray photoelectron spectra of pure tin oxide and gold/platinum loaded tin oxide aerogels, respectively. | ||
The high-resolution Pt 4f spectrum showed peaks with asymmetric shapes. A binding energy value of 71.2 eV found for the Pt 4f7/2 peak is characteristic for metallic platinum. The asymmetric shape, which resulted from numerous excited states, supported the assumption of the main presence of metallic platinum. The findings agreed with the studies by Elezovic et al. who also used the borohydride reduction method to obtain Pt particles supported on ATO. The authors also reported on the formation of mainly metallic platinum species.27 However, small differences between the recorded spectrum and the fitting curve observed at ca. 74.8 eV could result from moieties of oxidized platinum species. Oxidized species were analyzed on hybrid Pt–SnO2 materials produced by using borohydride as a reducing agent on carbon supports. Here, approximately half of the Pt was in oxidized states.28,29 Rizo et al. observed the formation of the intermetallic phase Pt3Sn but also a significant fraction of the oxidized Pt species supported on different carbon supports formed by coproduction with formic acid.30
The two Au 4f peaks appeared at 84.0 and 87.9 eV indicating that Au is in the metallic state. Due to the noble characteristic of Au, oxidized Au species are unlikely. Other reports on porous Au/SnO2 materials also show metallic Au particles, even though they have been calcined in air.30,31
CO oxidation
A tin oxide aerogel and two xerogels containing 5 wt% Au particles were tested by catalytic CO oxidation. As shown in the conversion plot of Fig. 10, the initial activity of the aerogel and the xerogel dried from hexane during the heat-up phase is lower than that of the xerogel. Most likely, this behavior can be explained by organic residues and CO2 from the supercritical drying process. These have to be removed from the aerogel or the xerogel (hexane) by heating first. At 200 °C, both, aerogel and xerogel (water) show a high CO conversion of 95%, and the xerogel (hexane) only reaches 75% conversion. After activation at 300 °C in O2/N2 stream the activity is further increased, resulting in nearly full conversion >97% at 200 °C for the aerogel and xerogel (water). The xerogel (hexane) shows a conversion of 96% but significantly increased its activity compared to 200 °C before activation. The actual performance of the catalyst is rated by the conversion at low temperature after the activation. The xerogel (water) shows the best results, with still 77% CO conversion at 50 °C. The aerogel and the xerogel (hexane) show similar, but lower conversions of 68% and 67%, respectively, at the same temperature stage. It seems that the better retention of the pore structure by supercritical drying or drying from hexane with low surface tension does not have a positive influence on the catalytic performance in this case. The CO molecule is rather small and diffusion to the active site is not the rate limiting step in this reaction. Unlike the xerogel (water) the aerogel and the xerogel (hexane) were in contact with organic solvents before drying. Therefore we assume that the contact with organic solvents or the residues adsorbed on the surface after drying caused the degradation of the gold surface. Unfortunately, there is no way to determine the specific Au surface area by TEM imaging, because even if the average diameter of the Au particles is known, the fraction of the particle surface covered by contact to the tin oxide support remains unknown.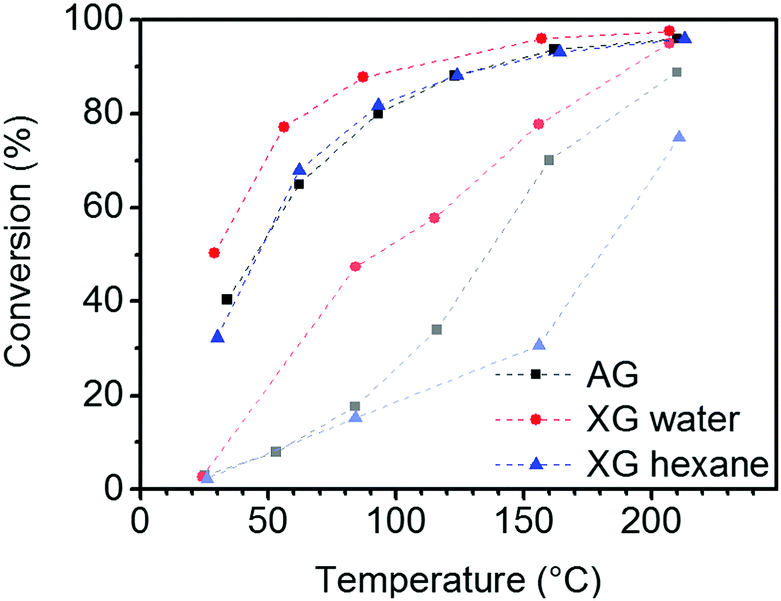 | ||
| Fig. 10 CO conversion in catalytic oxidation with 5% Au/SnO2 aerogel (AG) and xerogels (XG) (curves in light colors indicate the CO conversion during the heat-up phase). | ||
The catalytic performance of the Au/SnO2 gels does not compete with 2 nm small Au particles supported on TiO2 or Fe2O3 obtained by impregnation or a coprecipitation route. These catalysts reach full conversion even at low temperatures.32 The performance of the aerogel catalyst may be improved by using smaller Au particles for cogelation, but synthesis requires stronger ligands such as CTAB or organic media. Before cogelation the necessary ligand exchange or phase transfer without agglomeration of very small Au particles is challenging. Strong adsorbing amines or even thiol ligands may also block catalytic sites.
This work shows the use of mixed aerogels obtained by cogelation of preformed particles. By separate colloidal synthesis it is possible to form alloyed particles or particles with distinct facets. Using these in a cogelation like the one presented here allows the preparation of catalysts tailored to specific reaction that hardly can be obtained by classical coprecipitation or impregnation routes. Furthermore, it also may be used for tin oxide based sensor materials whose behavior and selectivity is enhanced by metal particles.
Conclusions
Tin oxide aerogels were synthesized by a facile, all-inorganic route starting with hexahydroxostannate precursors. The gels exhibit a sponge like morphology and high specific surface areas up to 375 m2 g−1.The synthesis route can be modified by adding hexahydroxoantimonate to obtain antimony-doped aerogels, which exhibit electrical conductivity after calcination in air at 550 °C. Samples containing 7.5 and 10 at% Sb exhibit the best conductivity and retain a specific surface area of up to 130 m2 g−1, which is the highest value for ATO aerogel samples calcined at this temperature.
Using this method it is also possible to obtain mixed gels by cogelation. In this, the gelation of the tin oxide sol is induced after mixing it with a sol of other nanoparticles, which was exemplarily shown for gold and platinum nanoparticles. The particles are evenly distributed over the gel without agglomeration. The good dispersion of the Au nanoparticles is shown by catalytic tests of CO oxidation, which is sensitive to the particle size.
To conclude, this synthesis method is very facile as it can be done in beakers at room temperature and avoids expensive or dangerous reagents. It can be easily modified to obtain conductive ATO aerogels or gels loaded with other nanoparticles.
Future work should center on applications such as catalysis or sensors that benefit from the large specific surface area and the transport properties that are attributed to the wide pore size distribution of the tin oxide aerogel.
Experimental
This section contains only a brief description of the actual tin oxide aerogel synthesis. For details on characterization and test series see the ESI.†In order to obtain tin oxide sol, 1.5 mmol nitric acid was added to a freshly prepared solution of 1 mmol sodium or potassium hexahydroxostannate in 40 mL of deionized water under vigorous stirring. The solution turns turbid but clears again after some minutes. The sol is stirred for one more hour. The sol can be destabilized to a solvogel by the addition of an organic solvent (ethanol) or a further addition of acid to a total amount of more than 2 eq. of H+ per stannate ion. The gels are washed several times with water and the solvent was exchanged to acetone. Supercritical drying was performed after the exchange of acetone against liquid CO2 in an autoclave and heating the closed autoclave to 40 °C. The CO2 becomes supercritical and is slowly released.
To obtain ATO aerogels, 2.5 to 10 mol% of potassium hexahydroxostannate was replaced by potassium hexahydroxoantimonate(V) prior to sol synthesis. While calcining ATO aerogels at 550 °C in a muffle furnace the gels turn blue.
Citrate-stabilized Au or Pt particles were synthesized following the route described by Brown et al.26 or Bigall et al., respectively.25 The noble metal particle sols were mixed with the tin oxide sol without any treatment and cogelation was induced by the addition of 3.5 mmol nitric acid per mmol hexahydroxostannate.
The obtained aerogels were characterized using powder X-ray diffraction, nitrogen physisorption, scanning and transmission electron microscopy and X-ray photoelectron spectroscopy.
For the 5% Au/SnO2 system also xerogels from water and hexane as solvents via conventional drying were produced and their catalytic performance in CO oxidation was tested.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the support from the European Research Council (ERC-2013-AdG 340419 AEROCAT). We also thank Christine Damm from IFW Dresden for helping with TEM measurements.Notes and references
- H. Yang, G. Yu and H. Liu, J. Electron. Mater., 2015, 44, 3744–3751 CrossRef CAS.
- L. Ding, S. He, S. Miao, M. R. Jorgensen, S. Leubner, C. Yan, S. G. Hickey, A. Eychmüller, J. Xu and O. G. Schmidt, Sci. Rep., 2014, 4, 4647 CrossRef PubMed.
- S. Das and V. Jayaraman, Prog. Mater. Sci., 2014, 66, 112–255 CrossRef CAS.
- A. Birkel, Y.-G. Lee, D. Koll, X. Van Meerbeek, S. Frank, M. J. Choi, Y. S. Kang, K. Char and W. Tremel, Energy Environ. Sci., 2012, 5, 5392 CAS.
- M. Davis, W. M. Hikal, C. Gümeci and L. J. Hope-Weeks, Catal. Sci. Technol., 2012, 2, 922–924 CAS.
- S. Vadivel, J. Mater. Sci.: Mater. Electron., 2015, 26, 7127–7133 CrossRef CAS.
- S. Ostrovsky, M. J. Larsen, A. Peled and J.-P. Lellouche, J. Nanopart. Res., 2015, 17, 273 CrossRef.
- G. Ozouf and C. Beauger, J. Mater. Sci., 2016, 51, 5305–5320 CrossRef CAS.
- E. Fabbri, A. Rabis, R. Kötz and T. J. Schmidt, Phys. Chem. Chem. Phys., 2014, 16, 13672 RSC.
- N.-L. Wu, L.-F. Wu, Y.-C. Yang and S.-J. Huang, J. Mater. Res., 1996, 11, 813–820 CrossRef CAS.
- R. Huang, L. Hou, B. Zhou, Q. Zhao and S. Ren, J. Non-Cryst. Solids, 2005, 351, 23–28 CrossRef CAS.
- G. Cognard, G. Ozouf, C. Beauger, G. Berthomé, D. Riassetto, L. Dubau, R. Chattot, M. Chatenet and F. Maillard, Appl. Catal., B, 2017, 201, 381–390 CrossRef CAS.
- J. H. Harreld, J. Sakamoto and B. Dunn, J. Power Sources, 2003, 115, 19–26 CrossRef CAS.
- T. F. Baumann, S. O. Kucheyev, A. E. Gash and J. H. Satcher, Adv. Mater., 2005, 1546–1548 CrossRef CAS.
- J. P. Correa Baena and A. G. Agrios, J. Phys. Chem. C, 2014, 118, 17028–17035 CAS.
- J. P. Correa Baena and A. G. Agrios, ACS Appl. Mater. Interfaces, 2014, 6, 19127–19134 CAS.
- F. Rechberger, R. Städler, E. Tervoort and M. Niederberger, J. Sol-Gel Sci. Technol., 2016, 80, 660–666 CrossRef CAS.
- S. Mishra, E. Jeanneau, S. Mangematin, H. Chermette, M. Poor Kalhor, G. Bonnefont, G. Fantozzi, S. Le Floch, S. Pailhes and S. Daniele, Dalton Trans., 2015, 44, 6848–6862 RSC.
- H. W. Wang, G. D. Xu, J. R. Zhang and X. Yin, Bull. Korean Chem. Soc., 2014, 35, 1999–2003 CrossRef CAS.
- R. Dittmann, E. Wintermantel and T. Graule, J. Eur. Ceram. Soc., 2013, 33, 3257–3264 CrossRef CAS.
- A. F. Hollemann, E. Wiberg and N. Wiberg, in Lehrbuch der Anorganischen Chemie, Walter de Gruyter & Co., Berlin, New York, 102nd edn, 2007 Search PubMed.
- N. Li, Q. Meng and N. Zhang, Particuology, 2014, 17, 49–53 CrossRef CAS.
- F. Rouqerol, J. Rouqerol and K. Sing, Adsorption by Powders and Porous Solids: Principles, Methodology and Applications, Academic Press, London, UK, 1999 Search PubMed.
- T. Nütz, U. zum Felde and M. Haase, J. Chem. Phys., 1999, 110, 12142–12150 CrossRef.
- N. C. Bigall, A. Herrmann, M. Vogel, M. Rose, P. Simon, W. Carrillo-Cabrera, D. Dorfs, S. Kaskel, N. Gaponik and A. Eychmüller, Angew. Chem., Int. Ed., 2009, 48, 9731–9734 CrossRef CAS PubMed.
- K. R. Brown, D. G. Walter and M. J. Natan, Chem. Mater., 2000, 12, 306–313 CrossRef CAS.
- N. R. Elezovic, V. R. Radmilovic, J. Kovac, B. M. Babic and L. M. Gajic-krstajic, RSC Adv., 2015, 5, 15923–15929 RSC.
- G. Lu, X. Ma, H. Yang and D. Kong, Int. J. Hydrogen Energy, 2015, 40, 5889–5896 CrossRef CAS.
- X. Li, J. Wei, Y. Chai and S. Zhang, J. Colloid Interface Sci., 2015, 450, 74–81 CrossRef CAS PubMed.
- R. Rizo, D. Sebastián, M. Jesús and E. Pastor, Appl. Catal., B, 2017, 200, 246–254 CrossRef CAS.
- J. Zhang, X. Liu, S. Wu, M. Xu, X. Guo and S. Wang, J. Mater. Chem., 2010, 20, 6453 RSC.
- M. Haruta, S. Tsubota, T. Kobayashi, H. Kageyama, M. J. Genet and B. Delmon, J. Catal., 1993, 144, 175–192 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qm00395a |
| This journal is © the Partner Organisations 2018 |