
From ZIF nanoparticles to hierarchically porous carbon: toward very high surface area and high-performance supercapacitor electrode materials†
Fangfang
Wang
,
Liangkui
Zhu
,
Ying
Pan
,
Zhan
Li
,
Pingping
Yang
,
Mingqiu
Song
,
Zhuangzhuang
Gao
,
Qianrong
Fang
 ,
Ming
Xue
* and
Shilun
Qiu
,
Ming
Xue
* and
Shilun
Qiu
State Key Laboratory of Inorganic Synthesis and Preparative Chemistry, Jilin University, Changchun 130012, P. R. China. E-mail: xueming@jlu.edu.cn
First published on 6th October 2018
Abstract
Porous carbon materials with a high surface area have attracted considerable attention for their potential application in electrochemical energy storage. In this study, a high-performance capacitive energy storage material based on hierarchically porous carbon was successfully prepared from a new nanoscale ZIF (zeolitic imidazolate framework) precursor, JUC160. The effects of the activating reagent KOH on the textural characteristics and supercapacitor performances of ZIF-derived porous carbons have been carefully evaluated. The JUC160-700 sample has a high surface area (SBET = 3253 m2 g−1), a hierarchical porous structure with micro-/mesopore frameworks and an appropriate degree of graphitisation, all of which are crucial for the enhancement of electrochemical performance. In electrochemical evaluation, JUC160-700 exhibits an ultra-high capacitance (386 F g−1 at 1 A g−1), good rate capability (71.8% retention at 20 A g−1) and long-term cycling stability (>99.9% over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles). This remarkable performance indicates that ZIF-derived porous carbon could be an ideal electrode material for advanced supercapacitors and other electrochemical energy storage devices.
000 cycles). This remarkable performance indicates that ZIF-derived porous carbon could be an ideal electrode material for advanced supercapacitors and other electrochemical energy storage devices.
Introduction
Supercapacitors, or electrochemical capacitors, have received growing attention as prospective energy storage devices because of their high power density, rapid charging–discharging capacity and long cycle life.1–5 Depending on the energy storage mechanism, supercapacitors can be classified as electrical double-layer capacitors (EDLCs) or pseudocapacitors.6–8 At the interface between an electrode and electrolyte, pseudocapacitors store energy on the basis of a rapid and reversible redox reaction, whilst EDLCs use the adsorption–desorption of electrolyte ions to store electrical energy.9–12 Unlike pseudocapacitors, EDLCs usually exhibit excellent cycling stability and electrical conductivity, which allow their commercial application in electrochemical energy storage systems. Porous carbons, as the most popular candidate electrode materials for EDLCs, have attracted great attention due to their high level of electrical conductivity, stable physicochemical properties and environmental friendliness, among other advantages.13–19In general, a large specific surface area offers the benefits of creating low-resistant channels, shortening the distance of ion transport and contributing greatly to the formation of an EDLC, whilst a rationally designed pore structure with well-interconnected hierarchical pores is expected to improve the specific capacitance and rate capability.20–23 To yield high-quality porous carbon materials, several efficient and easily accomplished approaches based on chemical activation have been proposed and investigated. Among the various chemical reagents available, potassium hydroxide (KOH) is the most widely used because it can simultaneously improve the specific surface area, microporosity and electrochemical performance of the generated porous carbon.24–27 The synthesis of layered graphene nanoribbons, derived from rod-shaped MOF-74 crystals by chemical KOH treatment and thermal transformation, has been reported. The graphene nanoribbons demonstrated specific capacitances of 193 F g−1 and 164 F g−1 at 10 mV s−1.28
Metal–organic frameworks (MOFs), with tunable porous structures, higher surface areas and inherent presence of heteroatoms, have been demonstrated as ideal precursors for supercapacitor electrode materials,29–37 ever since first reported by Xu et al.38 Recently, zeolitic imidazolate frameworks (ZIFs), a subfamily of metal organic frameworks (MOFs) generated from an assembly of transition metal ions (i.e., Zn(II), Co(II)) and N-rich imidazolate linkers, have attracted extensive interest.39–43 The introduction of N atoms into carbon frameworks has been demonstrated to enhance the specific capacitance and conductivity of porous carbon materials.44–47 Xu et al. successfully synthesised porous carbons using ZIF-8 as both the precursor and the template, introducing furfuryl alcohol as an additional precursor. The specific capacitance of the resultant C800 reached 188 F g−1 at a scan rate of 5 mV s−1.48 In 2014, Xu et al. reported a hierarchically porous 3D carbon framework by the assembly of microporous ZIF-8 particles for the first time using ultrasonication during its synthesis. The specific capacitance of the prepared AS-ZC-800 is as high as 251 F g−1 at the current density of 0.25 A g−1.49 Yamauchi et al. selectively prepared nanoporous carbon from a single ZIF-67 precursor by optimising the heating temperature, and the ZIF-derived carbon material reached a high specific capacitance of 272 F g−1 at 5 mV s−1.50Via co-carbonisation of ZIF-7 and a second carbon source, glucose, Cao et al. fabricated carbon-L-950 that possessed a capacitance value of 228 F g−1 at 0.1 A g−1.51 These ZIFs have been demonstrated as appropriate precursors to derive porous carbons for electrode materials in supercapacitor applications, but substantial challenges remain in the rational design of N-doped porous carbon materials to satisfy the requirements of a high specific capacitance and long-term cycling stability.
In this work, to further improve the capacitive performance of porous carbon electrode materials, we synthesised nitrogen-doped porous carbons with a very high surface area and hierarchical pore structure by KOH activation of carbonaceous precursors derived from a new ZIF. These N-doped porous carbons present very high surface areas as large as 3253 m2 g−1, an appropriate degree of graphitisation, and a hierarchical pore structure with a narrow micro-/mesopore distribution. Such unique features make these porous carbons efficient electrode materials for application in energy storage devices. Remarkably, the JUC160-700 material demonstrates an ultra-high capacitance value of 386 F g−1 at 1.0 A g−1, high rate capability (71.8% retention at 20 A g−1) and an extremely long cycling life with 99.9% capacity retention after 10000 cycles at 10 A g−1. These nanoporous carbons derived from the JUC-160 precursor exhibit capacitive properties superior to most reported carbon-based electrodes.
Experimental section
Materials
All the chemicals were acquired from commercial sources and used without any further purification. Benzimidazole and 2-methylbenzimidazole were purchased from Sigma-Aldrich with a purity of 98%, zinc acetate dehydrate (Zn(Ac)2·2H2O) was obtained from Sinopharm Chemical Reagent Co., Ltd, and N,N′-dimethylformamide (DMF) was obtained from West Long Chemical Co., Ltd.Room temperature synthesis of JUC160
Zn(Ac)2·2H2O (9 mmol) was dissolved in 120 mL DMF solution; then the solution was slowly added into 160 mL DMF with benzimidazole (10 mmol) and 2-methylbenzimidazole (8 mmol). The mixed solution turned milky quickly; then the solution was vigorously stirred for 12 h at room temperature. The resultant product was centrifuged and washed with DMF and methanol several times to remove the solvent. Finally, the JUC160 material was dried overnight in an oven and stored at 60 °C.Synthesis of porous carbon materials
First, the as-synthesized JUC160 powder was placed in a ceramic boat and transferred to a tube furnace. Flowing Ar atmosphere was used to purge the furnace at room temperature for 1 h to exclude air. Then the powder was annealed at 700 °C with a heating rate of 5 °C min−1 for 4 hours under Ar flow. The resultant carbon was further activated with KOH. The mixture of KOH/carbon was ground well in a mortar at a weight ratio of 3/1 and placed on a quartz boat, and then inserted into the quartz tube furnace with flowing argon to exclude air before increasing the temperature. Subsequently the temperature was increased to 600 °C, 700 °C, 800 °C, or 900 °C at 5 °C min−1 for 30 min and decreased to normal temperature naturally under Ar flow. The entire calcination process was performed under a flowing Ar atmosphere. The resultant porous carbons were washed thoroughly with HCl solution (1 M) and DI water to neutral, and then dried overnight at 80 °C. The final nanoporous carbon samples were denoted as JUC160-T, where T refers to the activation temperature, 600, 700, 800 and 900 °C.Characterization
Powder X-ray diffraction (PXRD) was performed with a Shimadzu LabX XRD-6000 diffractometer using Cu-Kα radiation (λ = 1.5418 Å) at 35 kV, 25 mA. Raman spectra were recorded with a LabRAM HR800 Raman spectrometer and the samples were excited with a 532 nm laser. Elemental analysis was conducted using a Vario EL cube element analyzer. The N contents of JUC160-600, JUC160-700, JUC160-800 and JUC160-900 were 3.48 wt%, 2.95 wt%, 1.99 wt% and 0.26 wt%, respectively (Table 1). X-ray photoelectron spectroscopy (XPS) spectra were recorded by using a ESCALAB250 spectrometer. The scanning electron microscopy (SEM) image was observed with a JEOS JSM-6510 system. Transmission electron microscopy (TEM) images were recorded by using a Gatan 794 CCD camera on a JEM-2100 microscope. N2 sorption analysis was performed by using a Quantachrome Autosorb-iQ MP gas sorptometer at 77 K. The obtained carbon materials were degassed using a turbo molecular vacuum pump at 200 °C for 10 h prior to analysis. The Brunauer–Emmett–Teller (BET) method was used to calculate the surface areas based on the nitrogen adsorption data. The pore size distribution according to the N2 adsorption isotherm was calculated by the Non-Localized Density Functional Theory (NLDFT) method.| Samples |
S
BETa (m2 g−1) |
V
totalb (cm3 g−1) |
V
microc (cm3 g−1) |
D
pored (nm) |
N (wt%) | C (F g−1) | C (F g−1) |
|---|---|---|---|---|---|---|---|
| a S BET estimated in the relative pressure from 0.05 to 0.20 bar, which gives the best linearity. b Total (micro- and meso-) pore volume at the relative pressure P/P0 = 0.99. c Cumulative micropore volume with pore size ≤2 nm, the values in brackets are the percentage of micropore volume to total pore volume. d The median pore size calculated by the Horvath–Kawazoe (HK) method. e The capacitance calculated from GCD at a current density of 1 A g−1. f The capacitance calculated from GCD at 20 A g−1. | |||||||
| JUC160-600 | 2464 | 1.31 | 0.67 (51) | 0.58 | 3.48 | 346 | 208 |
| JUC160-700 | 3253 | 2.12 | 0.69 (33) | 0.60 | 2.95 | 386 | 277 |
| JUC160-800 | 3028 | 2.41 | 0.53 (22) | 0.59 | 1.99 | 334 | 253 |
| JUC160-900 | 2594 | 2.16 | 0.43 (20) | 0.59 | 0.26 | 257 | 194 |
Electrochemical measurements
All electrochemical behavior was examined in a 6.0 M KOH electrolyte with a three-electrode system using Hg/HgO and platinum plate as the reference electrode and counter electrode, respectively. A homogeneous slurry of dispersed porous carbon, polytetrafluoroethylene (PTFE) (60 wt%, diluted before use to 10 wt% in water) and acetylene black in ethanol at a weight ratio of 8:1:1 was subsequently coated on 1 cm × 1 cm nickel foam under 10 MPa and dried overnight at 80 °C to prepare the working electrode. The typical mass loading was about 3–5 mg cm−2 of the fabricated porous carbon-based electrode. The electrochemical characterization was conducted on an electrochemical workstation (CHI660D, Shanghai Chenhua Instruments Co.). Cyclic voltammetry (CV) measurement and galvanostatic charge–discharge (GCD) measurement were performed on the potential range of −1.0–0 V. The frequency of the electrochemical impedance spectroscopy (EIS) measurement was in the range of 100000 to 0.01 Hz with an amplitude of 5 mV.
The specific capacitance was calculated from the CV measurement according to:
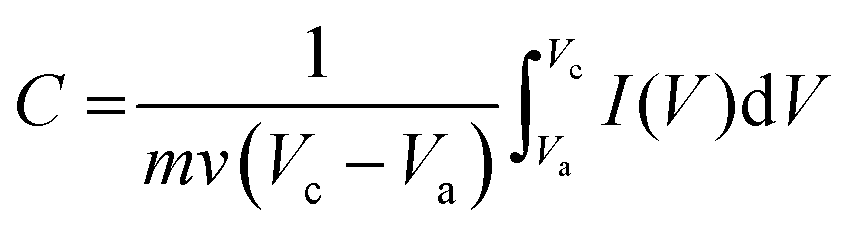 | (1) |
For galvanostatic measurement, the specific capacitance was calculated based on the following equation:
 | (2) |
Results and discussion
Structural evolution of nanoporous carbon materials
The JUC160 precursor (Zn4(2-mbIm)3(bIm)5·4H2O) was readily synthesised at room temperature. JUC160 is composed of four zinc metal centres, tetrahedrally coordinated by two large bulky ligand species, benzimidazole (bIm) and 2-methylbenzimidazole (2-mbIm), with a GIS (gismondine) zeolite topology (Fig. 1a). The powder X-ray diffraction (PXRD) pattern of the synthesised JUC160 was identical to the corresponding simulated XRD pattern (Fig. 1b), indicating phase purity with high crystallinity.52 Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images demonstrated that the JUC160 precursor exhibited a uniform cube-like morphology with average particle sizes of approximately 200 nm (Fig. 1c and d). | ||
| Fig. 1 (a) 3D structure of the JUC160 with the ZnN4 tetrahedron along the direction of [100] (Zn, dark green; N, sapphire blue; C, grey; H atoms and guest molecules both omitted for clarity); (b) PXRD of the simulated JUC160 (black), the as-synthesized JUC160 (red); (c) SEM and (d) TEM images of the as-synthesized JUC160. | ||
The JUC160 precursor was simply pretreated at 700 °C under an inert atmosphere and further subjected to KOH activation at 600 °C, 700 °C, 800 °C and 900 °C, separately. The resultant hierarchically nanoporous carbon samples were labelled as JUC160-T (where T refers to the activation temperature). These N-doped JUC160-T samples displayed similar diffraction features, with two broad and weak peaks located at around 2θ = 22° and 44° in the PXRD patterns, corresponding to the typical carbon (002) and (101) crystallographic facets, respectively, of graphitic carbon materials with a disordered orientation (Fig. 2a). Raman spectroscopy was performed to obtain further structural information on the JUC160-T samples. The intense G bands located at about 1590 cm−1 arose from the ordered graphitic layers, whilst the D bands observed at about 1342 cm−1 corresponded to the defective graphitic structures or partially disordered carbons (Fig. 2b). The degree of graphitisation was evaluated by using the intensity ratio between the D-band and G-band (ID/IG).53,54 The values of ID/IG for JUC160-600, JUC160-700, JUC160-800 and JUC160-900 were 0.97, 0.96, 0.98 and 1.07, respectively (Table S1†). The lowest ID/IG ratio was that of JUC160-700, demonstrating that it had the highest degree of graphitisation, which was expected to promote conductivity and rapid electron transport during the charge–discharge processes.
 | ||
| Fig. 2 (a) PXRD patterns and (b) Raman spectra of JUC160-T samples; (c) TEM and (d) HRTEM images of JUC160-700. | ||
The morphologies of the JUC160-T nanoporous carbons were observed via TEM. As shown in Fig. S1,† the pretreated precursors retained crystallite shapes similar to the JUC160 crystals with a certain amount of shrinkage and disintegration. Following KOH activation, Fig. 2c shows that the cube-like morphology of the JUC160 precursor was completely destroyed and that the annealed JUC160-700 presented an amorphous structure. The HRTEM image of JUC160-700 suggested the presence of abundant pores interconnected in a worm-like structure, which was expected to facilitate the fast diffusion of electrolyte ions (Fig. 2d). Meanwhile, TEM images also revealed that JUC160-600 had a distinctly compact appearance, whilst the internal structure of JUC160-900 was looser (Fig. S2†). These results confirm that the KOH activation process etched the carbon framework but eventually made the precursor morphology collapse and aggregate at higher temperatures due to excessive etching.
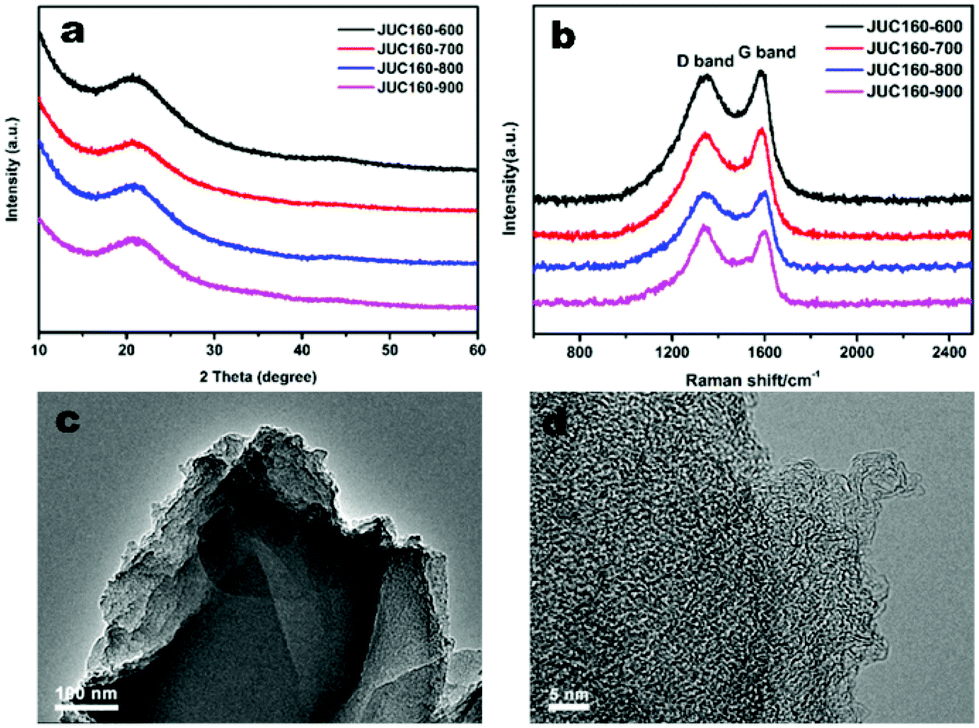
The porous structures of the JUC160-T samples were further evaluated by nitrogen adsorption–desorption experiments. The JUC160-700, JUC160-800 and JUC160-900 samples exhibited similar type I isotherms with a slight H4 hysteresis loop, and the JUC160-600 sample displayed a typical type I isotherm without such a loop, based on the IUPAC classification (Fig. 3a).52 The type I isotherms, characterised by a rapid nitrogen uptake at low relative pressure (P/P0 < 0.1), indicated that the porous carbons were endowed with abundant micropores. The hysteresis loop between adsorption and desorption branches confirmed the existence of mesopores. The pore size distributions of the prepared JUC160-T samples revealed the presence of micropores and some small mesopores with a size of 0.5 to 6 nm (Fig. 3b). The pore parameters and specific surface areas are summarised in Table 1. It is noteworthy that JUC160-700 exhibited the highest specific surface area of 3253 m3 g−1, a very high value relative to previously reported porous carbons. Importantly, we propose that the significantly increased specific surface area of JUC160-700 can be attributed to the efficient KOH activation at optimal temperature, whilst the slight decrease of the surface area from JUC160-700 to JUC160-900 was caused mainly by overactivation.25,55 Furthermore, the effects of pretreatment at temperatures of 650 °C and 750 °C were also investigated briefly. As shown in Fig. S3,† upon increasing the pretreatment temperature to 750 °C, the surface area was reduced to 2843 m2 g−1. Hence, the pretreatment temperature of 700 °C was the optimised condition to obtain porous carbons with a high surface area. In addition, compared to the low specific surface area of the precursor JUC-160 (210 m2 g−1), these porous carbons derived from JUC-160 had dramatically improved surface areas (Fig. S4†).
 | ||
| Fig. 3 (a) Nitrogen adsorption–desorption isotherms of JUC160-T at 77 K; (b) NLDFT pore size distribution curves of JUC160-T. | ||
In the well-known carbonisation process, the following reaction between carbon and KOH, as shown in eqn (3),56 occurs at low temperature (400–600 °C):
| 6KOH + 2C → 2K + 3H2 + 2K2CO3 | (3) |
Further reactions between K2O, K2CO3 and C (eqn (5) and (6)),25,57 the escape of CO2 and CO through the decomposition of K2CO3, and the subsequent decomposition of the as-formed K2CO3 into K2O and CO2 (eqn (4)) as the temperature increases to 700 °C all contribute to the development of porosity and a high specific surface area.
| K2CO3 → K2O + CO2 | (4) |
| K2CO3 + 2C → 2K + 3CO | (5) |
| K2O + C → 2K + CO | (6) |
When the temperature exceeds 700 °C, the excessive etching of the carbon framework by KOH severely damages the interconnected carbon walls, and the generated pore network collapses and aggregates.
The N contents of all four samples decreased as the calcination temperature increased, which was confirmed by the elemental analysis. The fact that higher calcination temperatures gave rise to higher specific surface areas while simultaneously liberating more nitrogen atoms implies a trade-off between surface area and N content. However, JUC160-700, synthesised at 700 °C, possessed both the highest specific surface area and a moderately high nitrogen content, thus simultaneously favouring its specific capacitance and rate capability.
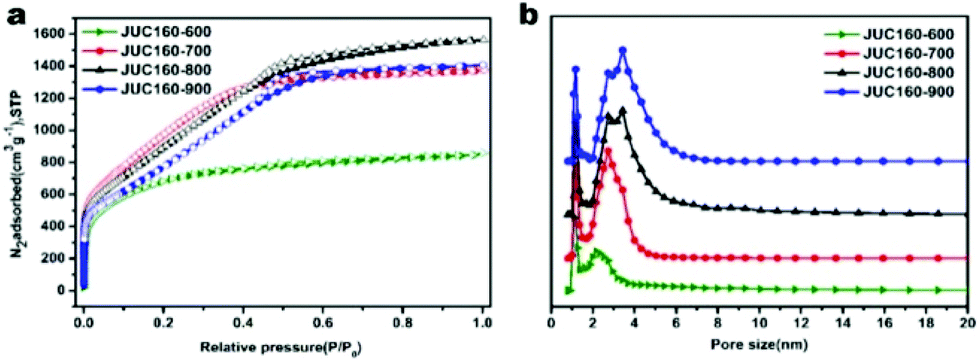
To further characterise the chemical states of the nitrogen atoms in the functionalised JUC160-700 sample, X-ray photoelectron spectroscopy (XPS) was carried out. As shown in Fig. 4a, the sample displayed typical characteristics of N-doped carbon materials, with the C 1s peaks for JUC160-700 centered at around 285.0 eV (corresponding to sp2 graphitic carbon) with some asymmetry.58 The high-resolution N 1s spectrum of JUC160-700 could be deconvoluted into four individual component peaks, which were assigned to pyridinic-N (397.8 eV), pyrrolic-N (400.2 eV), graphitic-N (401.2 eV) and pyridine-N-oxide (403.6 eV), respectively (Fig. 4b).59,60 The surface attachment of nitrogen atoms in various chemical states improved the conductivity of the porous carbons. On the basis of the above analysis, it is concluded that JUC160-700 reached the highest specific surface area, combined with a hierarchical porous structure, moderately high N content and appropriate graphitisation degree, all of which are indispensable prerequisites to achieve high-performance EDLCs.
 | ||
| Fig. 4 High-resolution XPS spectra of JUC160-700: (a) C 1s, (b) N 1s. | ||
Electrochemical characterisation of nanoporous carbons for EDLCs
The electrochemical performances of the nitrogen-doped JUC160-T samples were evaluated in a standard three-electrode system with the 6 M KOH electrolyte. All of the carbon electrode materials showed rectangular-like voltammograms in the potential range from −1 to 0 V at a sweep rate of 20 mV s−1, suggesting that the capacitive response arose mainly via a typical EDLC mechanism (Fig. 5a).61,62 Notably, reversible humps were observed in the quasi-rectangular cyclic voltammogram (CV) profiles, which demonstrated that the capacitive response of the materials arose via a combination of EDLC and faradaic reactions. The effect of rational N doping is known to induce pseudocapacitance and enhance the conductivity of the doped materials, thus contributing to the enhancement of their overall capacitive properties.63,64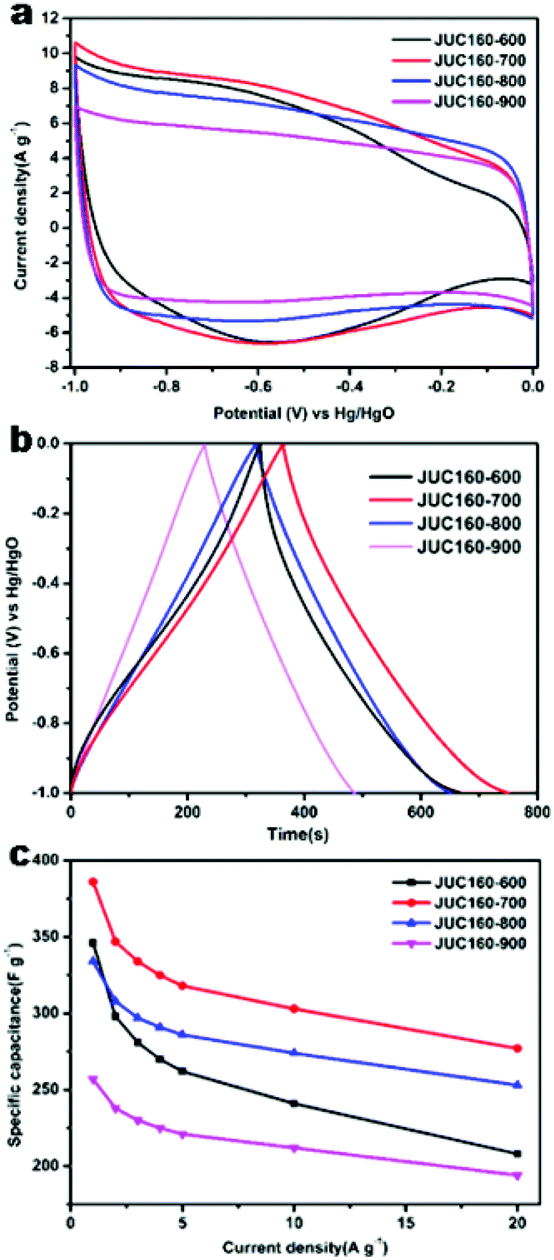 | ||
| Fig. 5 (a) Cyclic voltammetry curves of JUC160-T at 20 mV s−1; (b) galvanostatic charge–discharge profiles of JUC160-T at 1 A g−1; (c) specific capacitance versus different current densities of JUC160-T. | ||
A series of galvanostatic charge–discharge (GCD) measurements were conducted at 1 A g−1 to further assess the electrochemical performance of JUC160-T. As a result of the N doping effect, these porous carbon electrodes showed quasi-linear GCD curves with only a slight bend (Fig. 5b).65 As expected, JUC160-700 gave the largest capacitance value of 386 F g−1, which was much higher than that of JUC160-600 (346 F g−1), JUC160-800 (334 F g−1) and JUC160-900 (257 F g−1), at a current density of 1 A g−1. The specific capacitances of the porous carbon materials calculated from the GCD curves using eqn (2) are shown in Fig. 5c and Table S2.† The electrochemical behaviour of JUC160-700 was studied in thorough detail because it has the highest capacitance. As shown in Fig. 6a, with the sweep rates increasing from 5 to 200 mV s−1, the regular rectangular shape of the JUC160-700 curve showed no obvious distortion, suggesting that its hierarchically porous carbon structure led to superior capacitive behaviour. The CV curves of the other porous carbon materials, such as JUC160-600, JUC160-800 and JUC160-900, all indicated lower capacitance than JUC160-700 (Fig. S5a–S7a†). Using eqn (1), the capacitance of the JUC160-700 electrode was calculated as 329 F g−1 from the CV curve at 5 mV s−1 (Table S3†). GCD tests were also conducted to compare JUC160-700 with the other materials (Fig. 6b, Fig. S5b–S7b†). The isosceles shape of the JUC160-700 curve was well maintained, and the capacitance decreased only slightly to 277 F g−1 even at a high current density of 20 A g−1, which demonstrated the good coulombic efficiency and rate capability, consistent with the CV results.
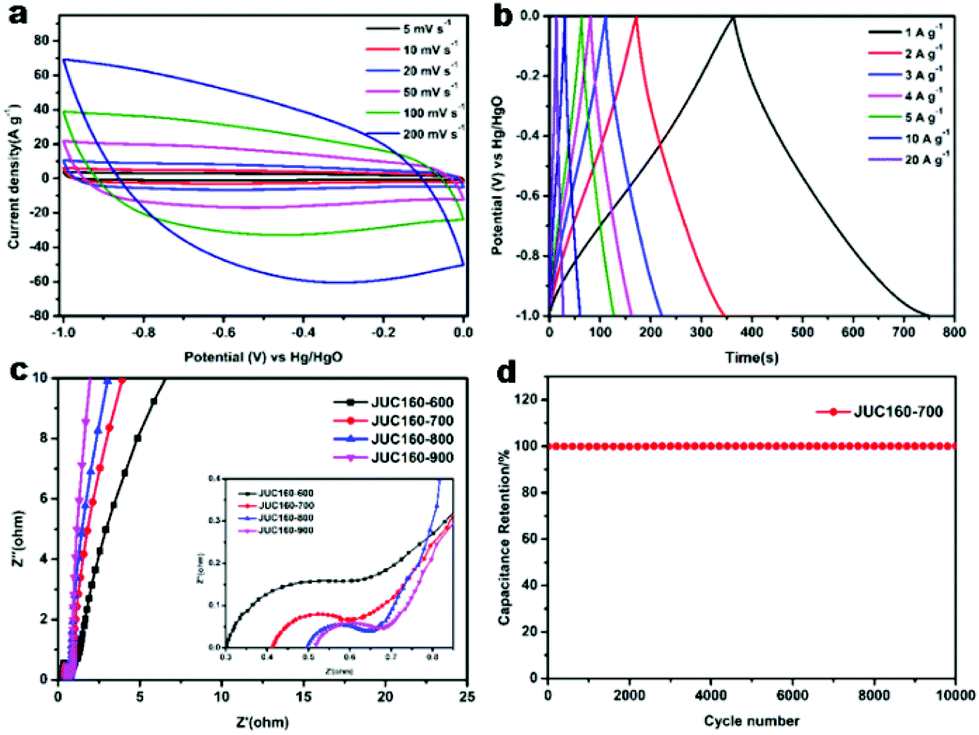 | ||
| Fig. 6 (a) Cyclic voltammetry profiles of JUC160-700 at different sweep rates; (b) galvanostatic charge–discharge curves of JUC160-700 at different current densities; (c) Nyquist plots of JUC160-T (frequency in the range of 105–10−2 Hz); (d) long-term cycling stability of the JUC160-700 at 10 A g−1. | ||
Compared with most previously reported porous carbons used as EDLC electrode materials, the JUC160-700 sample exhibits superior capacitive properties, which are ascribed to its high accessible surface area and well-interconnected micro-/mesoporous structure, which benefit the rapid transport of electrolyte ions and their diffusion to the electrode interface (Table 2).66 Electrochemical impedance spectroscopy (EIS) was then used to estimate the electrochemical performance and resistance of these porous carbons. The approximately vertical lines and small diameters of the semicircles in the low-frequency segment demonstrated that the JUC160-T samples had low internal resistance and charge–discharge resistance (Fig. 6c).76,77 The low resistance of the JUC160-T samples, which is consistent with the well-connected porous structures, revealed their favourable conductivity and capacitance characteristics.
| Carbon material | Precursor | S BET (m2 g−1) | Current densities/scan rates | Specific capacitance (F g−1) | Electrolyte | Ref. |
|---|---|---|---|---|---|---|
| JUC160-700 | JUC160 | 3253 | 1 A g−1/5 mV s−1 | 386/329 | 6 M KOH | This work |
| Carbon-L-950 | ZIF-7/glucose | 783 | 0.1 A g−1 | 228 | 6 M KOH | 51 |
| Large-size NPCs | ZIF-8 | 1523 | 5 mV s−1 | 251 | 1 M H2SO4 | 67 |
| CNT@CZIFs | ZIF-8/CNT | 287 | 0.5 A g−1 | 324 | 6 M KOH | 68 |
| MWCNT/NPC | ZIF-8/MWCNT | 928 | 2 A g−1 | 302 | 1 M H2SO4 | 69 |
| NC800-PEDOT | ZIF-8/PEDOT | 1186 | 5 mV s−1 | 218 | 1 M NaCl | 70 |
| Nano-PC | ZIF-67 | 350 | 5 mV s−1 | 272 | 6 M KOH | 71 |
| NC@GC | ZIF-8@ZIF-67 | 1276 | 2 A g−1 | 270 | 1 M H2SO4 | 64 |
| C-GMOF | MOF-5/GO | 979 | 2 mV s−1 | 345 | 6 M KOH | 72 |
| HPGCs | Ni(OH)2/resin | 970 | 1 A g−1 | 198 | 6 M KOH | 23 |
| 3D-HPCFs | GO-CNT@sponge | 1286 | 0.2 A g−1 | 379 | 6 M KOH | 73 |
| THPCs | Polypyrrole microsheets | 2870 | 0.5 A g−1 | 318 | 6 M KOH | 74 |
| CA-GA | D-Glucosamine | 571 | 0.1 A g−1 | 220 | 6 M KOH | 75 |
In practical applications, stability is also a crucial factor for supercapacitor electrode materials.78 The cycling stability of JUC160-700 was evaluated by GCD at a fairly high current density of 10 A g−1 (Fig. 6d). No obvious capacitance fading was noticed even after 10000 cycles, indicating the excellent cycling stability of the porous carbon. Clearly, JUC160-derived porous carbons show promise as efficient electrode materials owing to their superior electrochemical performance.
Conclusions
In summary, a new group of nitrogen-decorated hierarchically porous carbons have been successfully prepared using, as the precursor, nanoparticles of a new ZIF material (JUC-160) via an efficient KOH-activated route. The optimised material exhibits excellent supercapacitor performance, with an ultra-high specific capacitance of 386 F g−1 at 1.0 A g−1, and an extremely long cycle life, maintaining 99.9% capacity after 10000 cycles at 10 A g−1. This method could be easily adapted to fabricate other porous carbons with high surface area and good supercapacitive properties using ZIFs as precursors, and the development of advanced electrode materials is expected to be greatly accelerated by the increasing diversity of ZIFs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21571076, 21390394, 21571079) and the “111” project (B07016).Notes and references
- E. Lim, C. Jo and J. Lee, Nanoscale, 2016, 8, 7827–7833 RSC.
- H. Luo, Z. Liu, L. Chao, X. Wu, X. Lei, Z. Chang and X. Sun, J. Mater. Chem. A, 2015, 3, 3667–3675 RSC.
- B. Mendoza-Sanchez and Y. Gogotsi, Adv. Mater., 2016, 28, 6104–6135 CrossRef CAS.
- Y. Wang, Y. Song and Y. Xia, Chem. Soc. Rev., 2016, 45, 5925–5950 RSC.
- Z. Yu, L. Tetard, L. Zhai and J. Thomas, Energy Environ. Sci., 2015, 8, 702–730 RSC.
- S. Chen, W. Xing, J. Duan, X. Hu and S. Z. Qiao, J. Mater. Chem. A, 2013, 1, 2941–2954 RSC.
- W. Fan, C. Zhang, W. W. Tjiu, K. P. Pramoda, C. He and T. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 3382–3391 CrossRef CAS PubMed.
- M. Salanne, B. Rotenberg, K. Naoi, K. Kaneko, P. L. Taberna, C. P. Grey, B. Dunn and P. Simon, Nat. Energy, 2017, 1, 16070 CrossRef.
- S. Chen, M. Xue, Y. Li, Y. Pan, L. Zhu and S. Qiu, J. Mater. Chem. A, 2015, 3, 20145–20152 RSC.
- R. R. Salunkhe, Y. V. Kaneti, J. Kim, J. H. Kim and Y. Yamauchi, Acc. Chem. Res., 2016, 49, 2796–2806 CrossRef CAS.
- G. Yu, X. Xie, L. Pan, Z. Bao and Y. Cui, Nano Energy, 2013, 2, 213–234 CrossRef CAS.
- C. Yuan, H. B. Wu, Y. Xie and X. W. Lou, Angew. Chem., Int. Ed., 2014, 53, 1488–1504 CrossRef CAS.
- L. Dai, D. W. Chang, J. B. Baek and W. Lu, Small, 2012, 8, 1130–1166 CrossRef CAS.
- Y. Li, Z.-Y. Fu and B.-L. Su, Adv. Funct. Mater., 2012, 22, 4634–4667 CrossRef CAS.
- C. Liang, Z. Li and S. Dai, Angew. Chem., Int. Ed., 2008, 47, 3696–3717 CrossRef CAS.
- Y. Pan, Y. Zhao, S. Mu, Y. Wang, C. Jiang, Q. Liu, Q. Fang, M. Xue and S. Qiu, J. Mater. Chem. A, 2017, 5, 9544–9552 RSC.
- J. Xu, F. Xu, M. Qian, F. Xu, Z. Hong and F. Huang, Adv. Mater., 2017, 29, 1701674 CrossRef.
- G. Zhong, D. Liu and J. Zhang, J. Mater. Chem. A, 2018, 6, 1887–1899 RSC.
- L. Liu, Z. Niu and J. Chen, Chem. Soc. Rev., 2016, 45, 4340–4363 RSC.
- H. Itoi, H. Nishihara, T. Kogure and T. Kyotani, J. Am. Chem. Soc., 2011, 133, 1165–1167 CrossRef CAS.
- A. Mahmood, R. Zou, Q. Wang, W. Xia, H. Tabassum, B. Qiu and R. Zhao, ACS Appl. Mater. Interfaces, 2016, 8, 2148–2157 CrossRef CAS.
- C. Wang, C. Liu, J. Li, X. Sun, J. Shen, W. Han and L. Wang, Chem. Commun., 2017, 53, 1751–1754 RSC.
- D.-W. Wang, F. Li, M. Liu, G. Q. Lu and H.-M. Cheng, Angew. Chem., 2008, 120, 379–382 CrossRef.
- B. Ashourirad, A. K. Sekizkardes, S. Altarawneh and H. M. El-Kaderi, Chem. Mater., 2015, 27, 1349–1358 CrossRef CAS.
- J. Wang and S. Kaskel, J. Mater. Chem. A, 2012, 22, 23710–23725 RSC.
- X. Yang, M. Yu, Y. Zhao, C. Zhang, X. Wang and J.-X. Jiang, J. Mater. Chem., 2014, 2, 15139–15145 RSC.
- Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz and M. Thommes, Science, 2011, 332, 1537 CrossRef CAS.
- P. Pachfule, D. Shinde, M. Majumder and Q. Xu, Nat. Chem., 2016, 8, 718–724 CrossRef CAS.
- N. Ding, H. Li, X. Feng, Q. Wang, S. Wang, L. Ma, J. Zhou and B. Wang, J. Am. Chem. Soc., 2016, 138, 10100–10103 CrossRef CAS.
- B. Li, M. Chrzanowski, Y. Zhang and S. Ma, Coord. Chem. Rev., 2016, 307, 106–129 CrossRef CAS.
- Y. S. Meng, S. D. Jiang, B. W. Wang and S. Gao, Acc. Chem. Res., 2016, 49, 2381–2389 CrossRef CAS PubMed.
- S. Qiu, M. Xue and G. Zhu, Chem. Soc. Rev., 2014, 43, 6116–6140 RSC.
- H. Wang, Q.-L. Zhu, R. Zou and Q. Xu, Chem, 2017, 2, 52–80 CAS.
- J. P. Zhang, P. Q. Liao, H. L. Zhou, R. B. Lin and X. M. Chen, Chem. Soc. Rev., 2014, 43, 5789–5814 RSC.
- Z. Zhang, Z.-Z. Yao, S. Xiang and B. Chen, Energy Environ. Sci., 2014, 7, 2868–2899 RSC.
- A. Cadiau, K. Adil, P. M. Bhatt, Y. Belmabkhout and M. Eddaoudi, Science, 2016, 353, 137 CrossRef CAS.
- S. Dang, Q. L. Zhu and Q. Xu, Nat. Rev. Mater., 2017, 3, 17075 CrossRef.
- B. Liu, H. Shioyama, T. Akita and Q. Xu, J. Am. Chem. Soc., 2008, 130, 5390–5391 CrossRef CAS.
- R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Science, 2008, 319, 939 CrossRef CAS.
- S. Chen, M. Xue, Y. Li, Y. Pan, L. Zhu, D. Zhang, Q. Fang and S. Qiu, Inorg. Chem. Front., 2015, 2, 177–183 RSC.
- Y. Pan, K. Sun, S. Liu, X. Cao, K. Wu, W. C. Cheong, Z. Chen, Y. Wang, Y. Li, Y. Liu, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 2610–2618 CrossRef CAS.
- Y. Pan, M. Xue, M. Chen, Q. Fang, L. Zhu, V. Valtchev and S. Qiu, Inorg. Chem. Front., 2016, 3, 1112–1118 RSC.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS PubMed.
- L. F. Chen, X. D. Zhang, H. W. Liang, M. Kong, Q. F. Guan, P. Chen, Z. Y. Wu and S. H. Yu, ACS Nano, 2012, 6, 7092–7102 CrossRef CAS PubMed.
- L.-F. Chen, Y. Lu, L. Yu and X. W. Lou, Energy Environ. Sci., 2017, 10, 1777–1783 RSC.
- Z. Li, Z. Xu, X. Tan, H. Wang, C. M. B. Holt, T. Stephenson, B. C. Olsen and D. Mitlin, Energy Environ. Sci., 2013, 6, 871–878 RSC.
- F. Su, C. K. Poh, J. S. Chen, G. Xu, D. Wang, Q. Li, J. Lin and X. W. Lou, Energy Environ. Sci., 2011, 4, 717–724 RSC.
- H. L. Jiang, B. Liu, Y. Q. Lan, K. Kuratani, T. Akita, H. Shioyama, F. Zong and Q. Xu, J. Am. Chem. Soc., 2011, 133, 11854–11857 CrossRef CAS.
- A. J. Amali, J. K. Sun and Q. Xu, Chem. Commun., 2014, 50, 1519–1522 RSC.
- R. R. Salunkhe, J. Tang, Y. Kamachi, T. Nakato, J. H. Kim and Y. Yamauchi, ACS Nano, 2015, 9, 6288–6296 CrossRef CAS.
- P. Zhang, F. Sun, Z. Shen and D. Cao, J. Mater. Chem. A, 2014, 7, 442–450 CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051 CAS.
- A. C. Ferrari and D. M. Basko, Nat. Nanotechnol., 2013, 8, 235–246 CrossRef CAS.
- S. Gayathri, P. Jayabal, M. Kottaisamy and V. Ramakrishnan, AIP Adv., 2014, 4, 027116 CrossRef.
- E. Raymundo-Piñero, P. Azaïs, T. Cacciaguerra, D. Cazorla-Amorós, A. Linares-Solano and F. Béguin, Carbon, 2005, 43, 786–795 CrossRef.
- M. Armandi, B. Bonelli, F. Geobaldo and E. Garrone, Microporous Mesoporous Mater., 2010, 132, 414–420 CrossRef CAS.
- J. Romanos, M. Beckner, T. Rash, L. Firlej, B. Kuchta, P. Yu, G. Suppes, C. Wexler and P. Pfeifer, Nanotechnology, 2012, 23, 015401 CrossRef CAS.
- P. Zhang, F. Sun, Z. Xiang, Z. Shen, J. Yun and D. Cao, Energy Environ. Sci., 2014, 7, 442–450 RSC.
- J. W. Jeon, R. Sharma, P. Meduri, B. W. Arey, H. T. Schaef, J. L. Lutkenhaus, J. P. Lemmon, P. K. Thallapally, M. I. Nandasiri, B. P. McGrail and S. K. Nune, ACS Appl. Mater. Interfaces, 2014, 6, 7214–7222 CrossRef CAS.
- G. Zhang, L. Wang, Y. Hao, X. Jin, Y. Xu, Y. Kuang, L. Dai and X. Sun, Adv. Funct. Mater., 2016, 26, 3340–3348 CrossRef CAS.
- R. B. Rakhi, W. Chen, D. Cha and H. N. Alshareef, Adv. Energy Mater., 2012, 2, 381–389 CrossRef CAS.
- Y. Zhai, Y. Dou, D. Zhao, P. F. Fulvio, R. T. Mayes and S. Dai, Adv. Mater., 2011, 23, 4828–4850 CrossRef CAS PubMed.
- S. Maldonado, S. Morin and K. J. Stevenson, Carbon, 2006, 44, 1429–1437 CrossRef CAS.
- J. Tang, R. R. Salunkhe, J. Liu, N. L. Torad, M. Imura, S. Furukawa and Y. Yamauchi, J. Am. Chem. Soc., 2015, 137, 1572–1580 CrossRef CAS.
- E. Iyyamperumal, S. Wang and L. Dai, ACS Nano, 2012, 6, 5259–5265 CrossRef CAS.
- N. L. Torad, R. R. Salunkhe, Y. Li, H. Hamoudi, M. Imura, Y. Sakka, C. C. Hu and Y. Yamauchi, Chem, 2014, 20, 7895–7900 CrossRef CAS.
- R. R. Salunkhe, Y. Kamachi, N. L. Torad, S. M. Hwang, Z. Sun, S. X. Dou, J. H. Kim and Y. Yamauchi, J. Mater. Chem. A, 2014, 2, 19848–19854 RSC.
- L. Wan, E. Shamsaei, C. D. Easton, D. Yu, Y. Liang, X. Chen, Z. Abbasi, A. Akbari, X. Zhang and H. Wang, Carbon, 2017, 121, 330–336 CrossRef CAS.
- X. Li, C. Hao, B. Tang, Y. Wang, M. Liu, Y. Wang, Y. Zhu, C. Lu and Z. Tang, Nanoscale, 2017, 9, 2178–2187 RSC.
- Y. Li, J. Kim, J. Wang, N. L. Liu, Y. Bando, A. A. Alshehri, Y. Yamauchi, C. H. Hou and K. C. Wu, Nanoscale, 2018, 10, 14852–14859 RSC.
- R. R. Salunkhe, J. Tang, Y. Kamachi, T. Nakato, J. H. Kim and Y. Yamauchi, ACS Nano, 2015, 9, 6288–6296 CrossRef CAS.
- L. Wang, T. Wei, L. Sheng, L. Jiang, X. Wu, Q. Zhou, B. Yuan, J. Yue, Z. Liu and Z. Fan, Nano Energy, 2016, 30, 84–92 CrossRef CAS.
- B. You, J. Jiang and S. Fan, ACS Appl. Mater. Interfaces, 2014, 6, 15302–15308 CrossRef CAS.
- L. Qie, W. Chen, H. Xu, X. Xiong, Y. Jiang, F. Zou, X. Hu, Y. Xin, Z. Zhang and Y. Huang, Energy Environ. Sci., 2014, 6, 2497–2504 RSC.
- L. Zhao, L. Z. Fan, M. Q. Zhou, H. Guan, S. Qiao, M. Antonietti and M. M. Titirici, Adv. Mater., 2010, 22, 5202–5206 CrossRef CAS.
- W. Tian, H. Zhang, H. Sun, M. O. Tadé and S. Wang, Carbon, 2017, 118, 98–105 CrossRef CAS.
- S. Zhong, C. Zhan and D. Cao, Carbon, 2015, 85, 51–59 CrossRef CAS.
- C. Liu, J. Wang, J. Li, X. Hu, P. Lin, J. Shen, X. Sun, W. Han and L. Wang, J. Mater. Chem. A, 2016, 4, 11916–11923 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Electrochemical analysis, TEM, gas adsorption and additional figures. See DOI: 10.1039/c8qi00832a |
| This journal is © the Partner Organisations 2019 |