
Facile fabrication of CuS microflower as a highly durable sodium-ion battery anode†
 a
Xizheng
Liu
*a
a
Xizheng
Liu
*a
Abstract
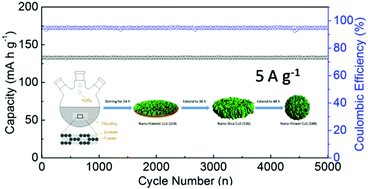
Sodium ion batteries (SIBs) have been considered as the promising substitutes for lithium ion batteries (LIBs) due to abundant resources of sodium. Metal sulfides have been demonstrated as prospective anode materials for SIBs based on a conversion mechanism. However, insufficient ionic transport and low conductivity in discharge electrodes prohibit their practical application. Herein, novel CuS microflowers were prepared by a facile dealloying method and applied as anode for SIBs. The microflowers are composed of nanosheets, which can provide increased Na+ diffusion admittance and more inter-space volume to accommodate volumetric change. When applied as anode in SIBs, the CuS microflowers-based anode delivered high discharge capacity (325.6 mA h g−1 at 0.1 A g−1) and excellent rate performance. The anode also displayed ultra-stable cycle performance and almost no capacity decay even after 5000 cycles (at a current density of 5 A g−1). Ex situ XRD was carried out to disclose the sodium ion storage mechanism. The CuS microflowers-based anode first experienced intercalation and then conversion mechanism with successive sodiation processes, while the reverse was observed for the charging process. The superior electrochemical performance is associated with the nano-micro structure and the controlled reaction mechanism. The current study states briefly that the simple and efficient dealloying method will provide more choices for fabricating anodes for SIBs.
- This article is part of the themed collection: Inorganic Chemistry Frontiers HOT articles for 2018
 Please wait while we load your content...
Please wait while we load your content...

