
A facile self-template and carbonization strategy to fabricate nickel nanoparticle supporting N-doped carbon microtubes†
 *a
*a
Abstract
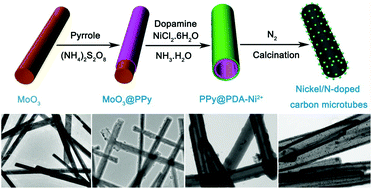
The efficient preparation of non-precious metal nanocatalysts embedded in carbon nano/microtubes remains a considerable challenge. Herein, we report a facile self-template and carbonization strategy to fabricate nickel nanoparticle (NP) supporting N-doped carbon microtubes. Firstly, molybdenum trioxide (a-MoO3) microrods are coated with polypyrrole (MoO3@PPy) by a simple in situ polymerization route. Then, PPy@PDA-Ni2+ microtubes with a hollow structure are obtained by further coating the PDA-Ni2+ complex in ammonia solution. In contrast, the direct coating of MoO3 microrods with PDA-Ni2+ under the same experimental conditions only leads to aggregated PDA-Ni2+ spheres. Moreover, nickel/N-doped carbon microtubes (Ni/NCMTs) can be obtained by a further heating treatment and highly graphitic carbon microtubes are achieved by etching the nickel nanoparticles. The hollow Ni/NCMTs exhibit excellent reduction activity on 4-nitrophenol (4-NP). Moreover, the size of Ni nanoparticles (Ni NPs) which can effectively control the catalytic performance of the as-prepared nanocomposites is facilely adjusted via changing the roasting temperature. Benefitting from the highly exposed surface, short diffusion distance, and homogeneous Ni NP dispersion, the Ni/NCMT catalyst exhibits an improved activity on 4-NP. This novel structure is helpful for further applications in hydrogen evolution reaction, supercapacitors and batteries.
- This article is part of the themed collection: Inorganic Chemistry Frontiers HOT articles for 2018
 Please wait while we load your content...
Please wait while we load your content...

