
A study on electrochemical properties of P2-type Na–Mn–Co–Cr–O cathodes for sodium-ion batteries
 *ab
Weiwei
Huang
*a
*ab
Weiwei
Huang
*a
Abstract
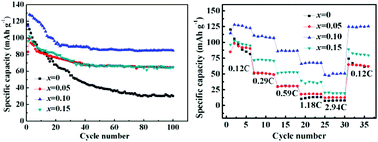
Na0.67Co0.25Mn0.75−xCrxO2 (x = 0, 0.05, 0.10, 0.15) oxides were prepared by a conventional solid-state reaction. The electrochemical properties of P2-type Na–Mn–Co–Cr–O cathodes for sodium-ion batteries have been investigated systematically. X-ray diffractometry and field emission scanning electron microscopy show that Cr ions are successfully incorporated into the lattice of the Na–Co–Mn–O oxide, and a P2-type layered structure with the P63/mmc space group remains unchanged after Cr-doping. The electrochemical measurements show that the maximum discharge capacity and the cycling stability of the matrix material is significantly enhanced by Cr-doping, which may be ascribed to the expanded a-axis and lattice volume, and the improvement of the rate capability is attributed to the significant decrease of the charge transfer resistance and the increase of the apparent Na+ diffusion coefficient. In the potential region of 2.0–4.0 V at 0.12C, Na0.67Co0.25Mn0.65Cr0.10O2 delivers the maximum discharge capacity of 128.1 mA h g−1, which is 10.31% higher than that of the un-doped sample, and the capacity retention of the sample is 66.35% after 100 cycles, which is 39.09% higher than that of the un-doped sample. It is suggested that the optimal Cr content can effectively improve the electrochemical properties of P2-type Na–Mn–Co–O cathodes for sodium ion batteries.
- This article is part of the themed collection: Inorganic Chemistry Frontiers HOT articles for 2018
 Please wait while we load your content...
Please wait while we load your content...

