
Facile preparation of monodisperse NiCo2O4 porous microcubes as a high capacity anode material for lithium ion batteries†
 *ab
and
Yongxing
Zhang
*a
*ab
and
Yongxing
Zhang
*a
Abstract
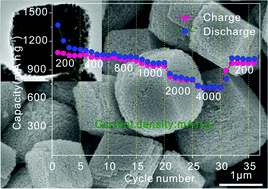
Binary transition metal oxides have attracted great attention as high-performance electrode materials for lithium-ion batteries in recent years. Herein, monodisperse NiCo2O4 porous microcubes were prepared for the first time via a simple urea-assisted solvothermal method followed by a thermal decomposition process. The porous microcubes assembled by nanoparticles with a size of ca. 35 nm have an average edge length of 1.5 μm. Nitrogen sorption isotherms show that this structure possesses a high surface area of 26.26 m2 g−1 with an average pore diameter of 22.57 nm. The rich mesopores among NiCo2O4 microcubes not only provide a large electrode/electrolyte reaction interface, but also provide enough void space to accommodate the volume change and prevent electronic disconnection in the electrode material during cycling. Furthermore, primary nanoparticles with a smaller size within microcubes can facilitate rapid Li-ion transport. So, when the as-prepared porous NiCo2O4 microcubes are used as anode materials for Li-ion batteries, they exhibit high-rate capability and outstanding cyclability.
- This article is part of the themed collection: Inorganic Chemistry Frontiers HOT articles for 2018
 Please wait while we load your content...
Please wait while we load your content...

