
A low temperature hydrothermal synthesis of delafossite CuCoO2 as an efficient electrocatalyst for the oxygen evolution reaction in alkaline solutions†
Zijuan
Du
 a,
Dehua
Xiong
*a,
Santosh Kumar
Verma
a,
Baoshun
Liu
a,
Xiujian
Zhao
a,
Lifeng
Liu
b and
Hong
Li
*a
a,
Dehua
Xiong
*a,
Santosh Kumar
Verma
a,
Baoshun
Liu
a,
Xiujian
Zhao
a,
Lifeng
Liu
b and
Hong
Li
*a
aState Key Laboratory of Silicate Materials for Architectures, Wuhan University of Technology, Wuhan 430070, P. R. China. E-mail: xiongdehua2010@gmail.com; lh_648@whut.edu.cn
bInternational Iberian Nanotechnology Laboratory (INL), Av. Mestre Jose Veiga, 4715-330 Braga, Portugal
First published on 13th November 2017
Abstract
Herein, we report the low temperature hydrothermal synthesis of delafossite CuCoO2 crystals at 100 °C. The structural, morphological and compositional characterization of CuCoO2 crystals was performed by powder X-ray diffraction (PXRD), field-emission scanning electron microscopy (FESEM) and X-ray photoelectron spectroscopy (XPS). Furthermore, the thermal stability of CuCoO2 in air and its electro-catalytic activity toward the oxygen evolution reaction (OER) have also been investigated.
Introduction
The discovery of new, economical and efficient catalysts for electrochemical energy conversion and storage is of prime importance to address the energy demand and the increasing concerns about environmental pollution. Among the electrochemical processes, the oxygen evolution reaction (OER) or water oxidation is the bottleneck of electrochemical water splitting (H2O → H2 + ½O2). Nowadays, noble metal oxides (IrO2 and RuO2) have been demonstrated to be the state-of-the-art electrocatalysts for the OER, but their high cost limits their large-scale industrial application. Therefore, lots of effort has been made in the exploration for efficient and low-cost catalytic materials comprising earth-abundant elements as alternatives to noble metal electrocatalysts.1,2From the 1970s, transition metal oxides such as ABO3 perovskite oxides (which are composed of rare and alkaline earth (A) and 3d transition metal cations (B)) have gained particular interest in an increasing number of scientific and industrial applications such as heterogeneous OER catalysts.3–8 A series of perovskite structure oxides such as ternary compounds LaNiO3,4 LaCoO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5 and quaternary compounds Ba0.5Sr0.5Co0.8Fe0.2O3−δ,6,7 and (Ln0.5Ba0.5)CoO3−δ (Ln = Pr, Sm, Gd and Ho)8 have been demonstrated to show an intrinsic activity comparable to the standard OER catalysts (IrO2 and RuO2). Besides, another kind of transition metal oxides such as delafossite oxides (the ternary oxides ABO2, where ‘A’ is a monovalent (Cu+ and Ag+) and ‘B’ is a trivalent (Al3+, Ga3+, In3+) cation) also have great potential as electro-catalysts and photo-catalysts for applications in electrochemical water splitting into hydrogen (H2) and oxygen (O2). A group of delafossite structure oxides such as CuRhO2, CuCoO2 and CuGaO2 have been reported to be promising candidates for OER electrocatalysis.9 Therefore, it is expected that these delafossite oxides could be useful for sustainable energy conversion applications.
5 and quaternary compounds Ba0.5Sr0.5Co0.8Fe0.2O3−δ,6,7 and (Ln0.5Ba0.5)CoO3−δ (Ln = Pr, Sm, Gd and Ho)8 have been demonstrated to show an intrinsic activity comparable to the standard OER catalysts (IrO2 and RuO2). Besides, another kind of transition metal oxides such as delafossite oxides (the ternary oxides ABO2, where ‘A’ is a monovalent (Cu+ and Ag+) and ‘B’ is a trivalent (Al3+, Ga3+, In3+) cation) also have great potential as electro-catalysts and photo-catalysts for applications in electrochemical water splitting into hydrogen (H2) and oxygen (O2). A group of delafossite structure oxides such as CuRhO2, CuCoO2 and CuGaO2 have been reported to be promising candidates for OER electrocatalysis.9 Therefore, it is expected that these delafossite oxides could be useful for sustainable energy conversion applications.
In the past few years, photo-electrochemical properties of delafossite oxides for solar cell applications were extensively studied,10–12 but only a few papers reported their applications in electrochemical water splitting. J. Ahmed and co-workers synthesized CuGaO2 nanoparticles from a sol–gel technique and used them as bifunctional electro-catalysts in O2 and H2 generation by the splitting of water.13 Reiko Hinogami and co-workers reported CuRhO2 delafossite as an active electrocatalyst for the OER in 1.0 M KOH electrolyte, and its cyclic voltammogram characteristic was comparable to that of Co3O4.14 J. Ahmed and co-workers investigated the electrocatalytic (OER) properties of CuGaO2, and found that the nanocrystalline CuGaO2 hexagons show an enhanced electrocatalytic activity compared to sub-micron-sized CuGaO2 plates and micron-sized CuGaO2 particles.15 Besides these studies on CuRhO2 and CuGaO2, there have been very few reports on the electro-catalysis of delafossite oxides, e.g. CuCrO2,9 CuFeO2,16 CuScO217 and CuMnO2.18 Copper based delafossite oxides have demonstrated excellent electronic properties but their electrochemical studies are still lacking, which motivates us to study delafossite oxides regarding their electrochemical performance for the OER or HER (hydrogen evolution reaction).
Generally, copper based delafossite oxides can be obtained through solid state reactions or sol–gel techniques, but the high reaction temperature (900–1000 °C) usually results in crystals with a large micro-meter size.19 The low temperature synthesis of delafossite oxides with controlled particle shape and size has been challenging. In our earlier studies, nano-sized delafossite oxides as active photoelectrode materials for solar cell devices such as CuCrO2,20–22 CuAlO2,23 CuGaO2,24 and AgCrO225 were obtained through a low temperature hydrothermal method at around 200 °C. More recently, we developed a facile hydrothermal route to synthesize CuFeO2 and CuMnO2 nanocrystals at 80–100 °C.26,27 In this work, we further report the preparation and electro-catalysis application of CuCoO2 crystals, which are also obtained through the low temperature hydrothermal method at 100 °C. To the best of our knowledge, there are only very few papers published on the preparation and properties of CuCoO2 crystals via the solid-state ion exchange (metathesis) reaction at 590 °C for 2 days,28,29 and the hydrothermal reaction at 210 °C for 60 hours,9,30 and these previous studies were unable to explicitly describe the crystal size and morphology and the resulting product contains an unknown phase9 or impurity of Co3O4.30 The present work displays the preparation and electrocatalytic application of CuCoO2 crystals. The crystal phase, morphology, composition and chemical states of elements, and the thermal stability and the OER performance of CuCoO2 crystals have been comprehensively studied. The as-fabricated GC@CuCoO2 electrode (GC@CuCoO2, the glassy carbon electrode (GC) supported CuCoO2 electrode) with an optimal loading of CuCoO2 catalysts exhibits efficient catalytic activity for the OER in alkaline solutions, requiring a low overpotential of 440 mV to attain an anodic current density of 10 mA cm−2 and showing fast kinetics for the OER with a small Tafel slope (92.8 mV dec−1) and charge transfer resistance.
Experimental details
Materials synthesis
All of the chemicals in these experiments were purchased from Sigma Aldrich, were of analytical gradeand used as received; CuCoO2 crystals were prepared according to our previous studies.26,27 Typically, 15 mmol Cu(NO3)2·3H2O and 15 mmol Co(NO3)2·6H2O were dissolved in 70 ml deionized water at room temperature; 4.40 g NaOH was added to the above solution and stirred for 10–20 minutes. And then, the solution was loaded into a 100 ml Teflon-lined autoclave. After the reaction was kept at 100 °C for 24 h, the obtained gray precipitate was washed with deionized water and absolute alcohol in sequence several times, and then stored in absolute alcohol solution.Structural characterization
The morphology, microstructure, and chemical composition of CuCoO2 were examined by field-emission scanning electron microscopy (FESEM, FEI Quanta 650) equipped with energy-dispersive X-ray spectroscopy (EDX). The crystalline structure of samples was studied by X-ray diffractometry (XRD, PANalytical X'Pert PRO) using Cu Kα radiation (λ = 1.540598 Å) and a PIXcel detector. The surface chemical states were analyzed by X-ray photoelectron spectroscopy (XPS, VG Multilab 2000). The thermal stability of CuCoO2 powders was investigated using a differential scanning calorimetry–thermogravimetric analysis (DSC-TG) device (Diamond TG/DTA, Perkin-Elmer Instruments); these samples were heated in air from room temperature to 800 °C at a heating rate of 10 °C min−1.Electrochemical measurements
The OER performance was evaluated by cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) in a three-electrode configuration in 1.0 M KOH (pH = 13.5) using a Biologic VMP-3 potentiostat/galvanostat. A platinum wire and a saturated calomel electrode (SCE) were used as the counter and reference electrodes, respectively. The working electrodes of GC@CuCoO2 were fabricated as follows. CuCoO2 powders were dispersed in a mixture of 500 μL water, 480 μL isopropanol and 20 μL Nafion (5 wt%, Sigma), and cast onto a piece of the GC electrode (CuCoO2 loading mass: 0.1, 0.2 and 0.3 mg cm−2). Cyclic voltammetric (CV) scans were recorded between 1.25 and 1.85 V vs. reversible hydrogen electrode (RHE) at the scan rate of 5 mV s−1. The electrochemical double-layer capacitance (Cdl) of each sample was measured given that Cdl is positively proportional to the effective surface areas (ESA). Cdl can be extracted through CV scans at different rates (from 10 to 100 mV s−1) in the non-faradaic potential window of −0.05 to 0.05 V vs. SCE. The EIS measurements were performed in the frequency range of 20 mHz–200 kHz under a constant potential of 1.60 V vs. RHE.All current density values are normalized with respect to the geometrical surface area of the working electrode. All CV curves presented in this work are iR-corrected. The correction was done according to the following equation:
| Ec = Em − iRs, | (1) |
| E(RHE) = E(SCE) + 0.241 + 0.059 pH. | (2) |
Results and discussion
Fig. 1a shows a SEM (scanning electron microscopy) image of the hydrothermal reaction products, where the delafossite feature of the hexagonal structure can be clearly seen. A close inspection reveals that the size of these crystals is around 1–2 μm, and the thickness of the lamellar crystal is around 300–400 nm (the inset in Fig. 1a). The XRD (X-ray diffraction) patterns of the reaction products can be indexed as CuCoO2 (JCPDS card No. 21-0256) from Fig. 1b, as evidenced by the characteristic diffraction peaks located at 15.5°, 31.5°, 36.9°, 37.7°, 39.6°, 42.1°, 57.2°, 61.8°, 65.1°, 66.8° and 74.2°, respectively, and no impurity phase was detected. It is worth mentioning that we, for the first time, obtained the pure CuCoO2 phase through the low temperature (100 °C) hydrothermal method, which is advantageous against the drawbacks of the high temperature hydrothermal reaction (e.g. 210 °C)9,30 by which impurities are usually inevitably generated. | ||
| Fig. 1 SEM images (a) and XRD patterns (b) of hydrothermally synthesized products. The inset in (a) is a high magnification SEM image. | ||
To analyze the chemical composition of CuCoO2, SEM-EDX (scanning electron microscopy-energy-dispersive X-ray spectroscopy) mapping was employed to characterize the powders. Fig. 2a shows a typical selected area for EDX mapping measurement, and the EDS analysis (Fig. 2e) indicates that the sample contains the elements Co, Cu and O. Moreover, the C originates from carbon tapes, which was applied to hold CuCoO2 powders. Furthermore, it can be observed that all of the Cu, Co and O elements are homogeneously distributed from Fig. 2b–d. In addition, the elemental chemical states of the CuCoO2 crystals have been investigated by XPS (X-ray photoelectron spectroscopy). The survey spectrum again confirms the presence of Cu and Co in freshly prepared CuCoO2 samples (Fig. S1, ESI†). The peaks located near 932.4 and 952.1 eV (Fig. 2f) correspond to the binding energies of Cu 2p3/2 and Cu 2p1/2, which were similar to those of CuCrO2, CuFeO2 and CuMnO2 in the Cu-based delafossite family with monovalent Cu ions. Besides, there are two shakeup satellites (identified as “Sat.”) in the Co 2p spectrum, and the other peaks close to 779.9 and 794.9 eV (Fig. 2g) represent the binding energies of Co 2p3/2 and Co 2p1/2, which were in agreement with the reported values of Co3+ in Co2O3 and Co3Se4.31–33 Moreover, the main peak close to 530.0 eV (Fig. 2h), resembling the binding energy of O 1s, suggests the metal–oxygen bonding of M–O.34 The weak peak located around 531.5 eV could be identified as H2O, which originated from absorbed water on the surface of the sample.34 Therefore, the monovalent state of the copper cation (Cu+) and the trivalent state of the cobalt cation (Co3+) in the samples can be explicitly confirmed.
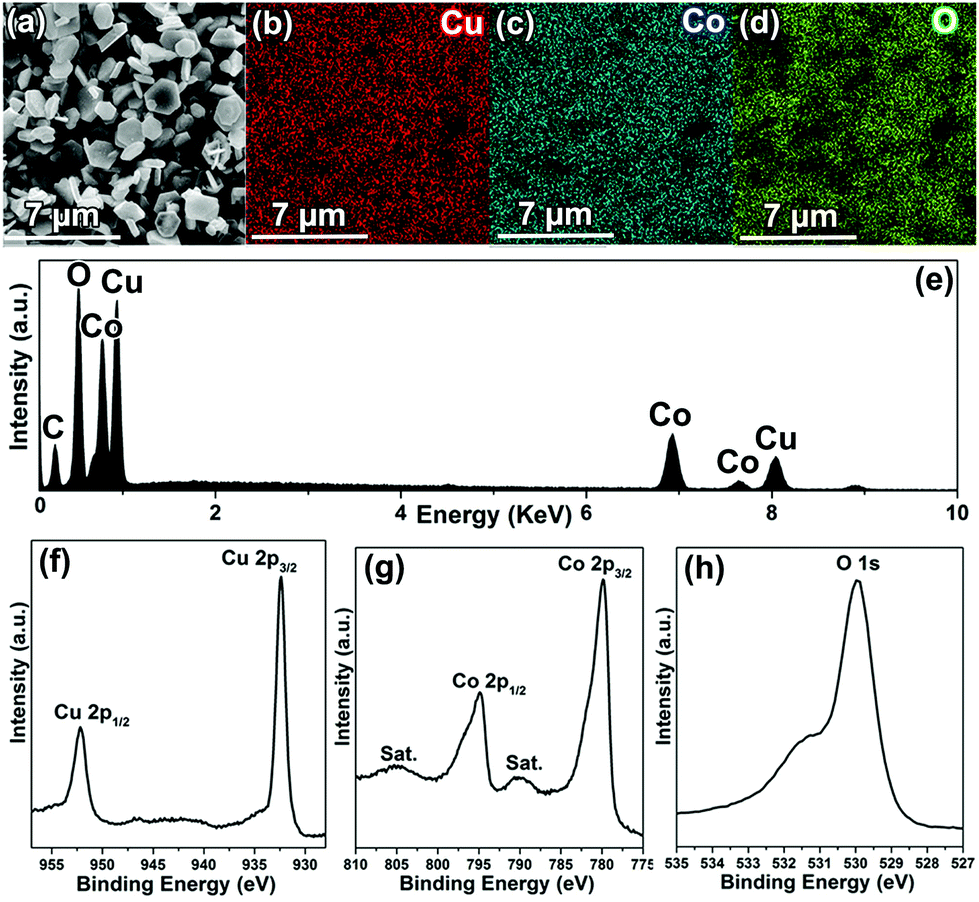 | ||
| Fig. 2 SEM image (a), elemental mapping (b, Cu; c, Mn; d, O), EDS spectrum (e) and XPS spectra (f, Cu 2p; g, Co 2p; h, O 1s) of CuCoO2 crystals. | ||
From the TG (thermogravimetric) curve shown in Fig. 3a, the initial weight loss may be caused by the evaporation of absorbed water from the CuCoO2 powders. The mass of CuCoO2 increases sharply above 500 °C in air, which should be due to the oxidation of CuCoO2; for example, monovalent copper (Cu+) was oxidized into divalent copper (Cu2+). The TG result of CuCoO2 is analogous to other copper based delafossite oxides reported before, e.g. CuAlO2,23 CuCrO2,20 CuGaO235 and CuMnO2.27 The XRD patterns (Fig. 3b) of CuCoO2 sintered at different temperatures (e.g. 300 and 600 °C) support our analysis. There is no remarkable difference for diffraction patterns between the CuCoO2 powders before and after sintering at 300 °C, and all these peaks can be identified as the pure CuCoO2 (JCPDS card No. 21-0256) phase. However, the diffraction pattern of CuCoO2 powders sintered at 600 °C is distinctly different from that of the as-prepared CuCoO2, and can be indexed as a mixture of the CuCo2O4 phase (JCPDS card No. 01-1155) and CuO phase (JCPDS card No. 45-0937), suggesting that the oxidization reaction of CuCoO2 has been completed. These results are consistent with the SEM images shown in Fig. 3c and d, where the morphology of these CuCoO2 crystals has an appearance similar to nanoplates, but the surface of CuCoO2 crystals melts after sintering at 600 °C (Fig. 3d), maybe owing to the newly generated copper cobalt composite oxide of CuCo2O4 and CuO. Thus, it is suggested that the following chemical reaction should be involved during the high-temperature (600 °C) sintering:
| 4CuCoO2 + O2 = 2CuCo2O4 + 2CuO. | (3) |
 | ||
| Fig. 3 (a) Thermogravimetric (TG) curves of CuCoO2 at a heating rate of 10 °C min−1 in air. Also shown are the corresponding XRD patterns (b) and SEM images of CuCoO2 powders after sintering at different temperatures (c, 300 °C; d, 600 °C) in air for 1 hour. | ||
The electrocatalytic activity of CuCoO2 towards the OER was evaluated in O2-saturated 1.0 M KOH solution using cyclic voltammetry (CV). The glassy carbon (GC) supported CuCoO2 electrode (i.e., GC@CuCoO2) was prepared using a method modified according to our previous report.34Fig. 4a shows the CV curves of GC electrodes with different loading masses of CuCoO2. For comparison, the OER activity of bare GC was also measured. The bare GC shows only a very small anodic current up to 1.70 V vs. RHE (reversible hydrogen electrode). After loading with CuCoO2 crystals, the anodic current densities of electrodes (GC@CuCoO2) are remarkably improved. From the reduction branch of the CV curves, GC@CuCoO2-0.1 mg shows a low onset potential (the potential at which the anodic current density is 1 mA cm−2) of 1.64 V vs. RHE (i.e., ηon = 410 mV), and this can reach 10 mA cm−2 at 1.73 V vs. RHE (i.e., η10 = 500 mV, the inset in Fig. 4a). GC@CuCoO2 with a high loading mass of CuCoO2 (such as 0.2 or 0.3 mg CuCoO2) exhibits better OER activity compared to that of GC@CuCoO2-0.1 mg, which shows a lower onset potential of 1.62 V vs. RHE (i.e., ηon = 390 mV), and can reach 10 mA cm−2 at 1.67 V (i.e., η10 = 440 mV) for the GC@CuCoO2-0.3 mg electrode. The small difference in the overpotential (ηon, η10) for these three samples is around 10–30 mV, indicating that a higher loading of CuCoO2 does not help to further boost the OER activity of the electrode. The OER activity of these GC@CuCoO2 electrodes is comparable to or better than many other oxide based OER catalysts recently reported in the literature (see Table S1, ESI†), such as perovskite oxide catalysts (LaNiO3 (η0.12 ≈ 400 mV),4 LaCoO3 (80 nm, η10 = 490 mV),5 Ba0.5Sr0.5Co0.8Fe0.2O3−δ (η10 = 370 mV),6 LaFeO3 (η10 = 510 mV),36 SrCoO3 (η0.7 ≈ 350 mV),37 SrCoO3−δ (η3 ≈ 320 mV),38 Pr0.5Ba0.5Co2O3−δ (η0.3 ≈ 320 mV)38) or other delafossite oxide catalysts (micron-sized CuRhO2 particles (η10 ≈ 420 mV),9 sub-micron-sized CuCoO2 plates (η10 ≈ 430 mV),9 micron-sized CuFeO2 plates (η5 ≈ 670 mV),9 micron-sized CuCrO2 plates (η5 ≈ 710 mV),9 CuGaO2 nanoparticles (η18 ≈ 370 mV),13 micron-sized CuRhO2 plates (η0.1 ≈ 380 mV),14 micron-sized CuGaO2 particles (η5 ≈ 370 mV),15 and sub-micron sized CuGaO2 plates (η23 ≈ 370 mV)).15
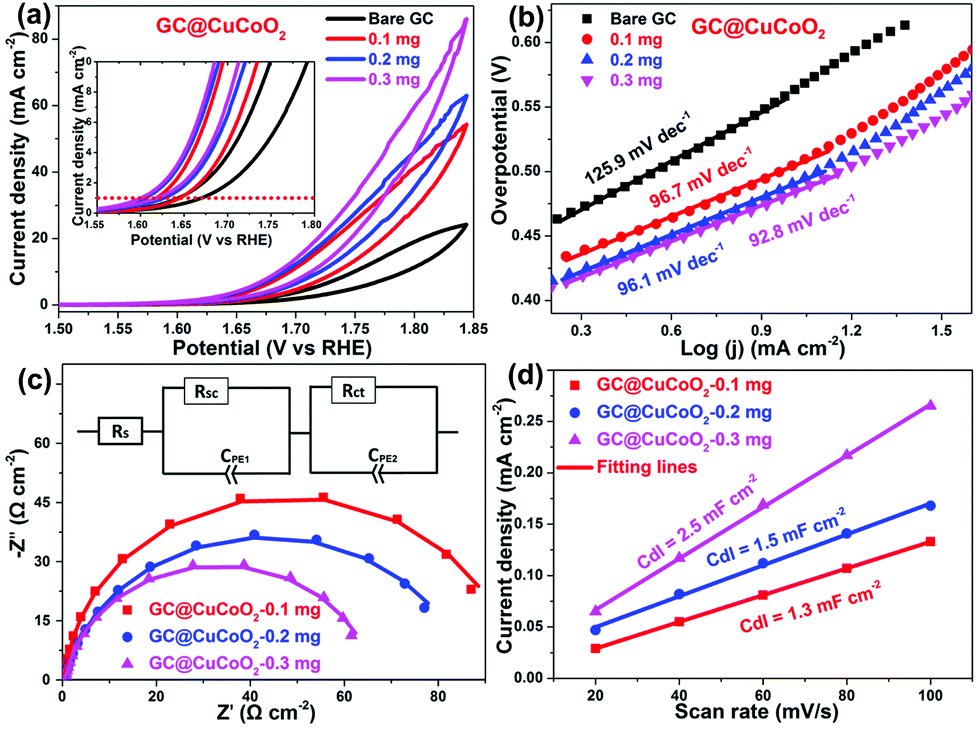 | ||
| Fig. 4 (a) CV curves and (b) Tafel plots of the bare GC and GC@CuCoO2 electrodes, (c) Nyquist plots of the GC@CuCoO2 electrodes, the inset of (c): equivalent circuit model. (d) Half of the difference between anodic and cathodic current densities (ΔJ = (Ja − Jc)/2) at 0 V vs. SCE plotted against the scan rate. The linear slope is equivalent to the double-layer capacitance Cdl that is proportional to the ECSA of GC@CuCoO2 electrodes. All measurements were performed in 1.0 M KOH at room temperature (ca. 25 °C). | ||
To gain additional understanding of the OER process, Tafel analysis was carried out in the linear potential region by fitting the reduced branches of the CV curves with the Tafel equation.33 The Tafel slope is found to be 125.9, 96.7, 96.1 and 92.8 mV dec−1 for the bare GC, GC@CuCoO2-0.1 mg, GC@CuCoO2-0.2 mg and GC@CuCoO2-0.3 mg electrodes, respectively (Fig. 4b). The Tafel slope of GC@CuCoO2 electrodes is substantially lower than that of many oxide-based OER catalysts (Table S1, ESI†), suggesting much faster kinetics towards the OER. This was also corroborated by our EIS study. Fig. 4c displays the EIS Nyquist plots of the bare GC and GC@CuCoO2 electrodes, which are fitted with the equivalent circuit model shown in the inset of Fig. 4c. According to the fitting results (Table S2, ESI†), the charge transfer resistance (Rct) of GC@CuCoO2-0.3 mg electrodes is 65.4 Ω cm−2, smaller than that of the GC@CuCoO2-0.2 mg (85.2 Ω cm−2) or GC@CuCoO2-0.1 mg (87.9 Ω cm−2), the Rct values of GC@CuCoO2 electrodes are smaller than those of other oxide catalysts, such as LaCoO3 (at the potential of 1.67 V vs. RHE)5 and LaFeO3 (at the potential of 1.65 V vs. RHE).36 These results indicate that the CuCoO2 crystals, as an efficient OER catalyst, can dramatically accelerate the OER process. The electrochemical double-layer capacitance (Cdl) of all the GC@CuCoO2 electrodes were measured to compare the effective surface areas (ESA), given that Cdl is positively proportional to ESA; a larger Cdl represents a higher surface area of the investigated catalyst. Cdl was measured through CV scans (Fig. S2–S4, ESI†) in the non-faradaic potential region (−0.05 to 0.05 vs. SCE) at different scan rates (20, 40, 60, 80, 100 mV s−1). As shown in Fig. 4d, the Cdl values of the GC@CuCoO2-0.1 mg, GC@CuCoO2-0.2 mg and GC@CuCoO2-0.3 mg electrodes are 1.3, 1.5 and 2.5 mF cm−2, respectively. The ESA was calculated from the Cdl according to the following equation:
| ESA = Cdl/Cs, | (4) |
Conclusions
In summary, we have synthesized delafossite CuCoO2 through a hydrothermal method at a low temperature (100 °C) for the first time. CuCoO2 supported on GC electrodes exhibits an efficient catalytic activity for the oxygen evolution reaction in alkaline solutions. Given the low-cost nature and ease of up-scale fabrication of electrodes through the low temperature hydrothermal method, delafossite CuCoO2 holds substantial promise for use as an efficient and inexpensive OER catalyst in water electrolyzers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to express sincere thanks for financial support by the National Natural Science Foundation of China (No. 51402223, 51772224).Notes and references
- M. Tahir, L. Pan, F. Idrees, X. Zhang, L. Wang, J.-J. Zou and Z. L. Wang, Nano Energy, 2017, 37, 136–157 CrossRef CAS.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- S. Gupta, W. Kellogg, H. Xu, X. Liu, J. Cho and G. Wu, Chem. – Asian J., 2016, 11, 10–21 CrossRef CAS PubMed.
- J. Bak, H. B. Bae, J. Kim, J. Oh and S.-Y. Chung, Nano Lett., 2017, 17, 3126–3132 CrossRef CAS PubMed.
- S. Zhou, X. Miao, X. Zhao, C. Ma, Y. Qiu, Z. Hu, J. Zhao, L. Shi and J. Zeng, Nat. Commun., 2016, 7, 11510 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- K. J. May, C. E. Carlton, K. A. Stoerzinger, M. Risch, J. Suntivich, Y.-L. Lee, A. Grimaud and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 3264–3270 CrossRef CAS.
- A. Grimaud, K. J. May, C. E. Carlton, Y.-L. Lee, M. Risch, W. T. Hong, J. Zhou and Y. Shao-Horn, Nat. Commun., 2013, 4, 2439 Search PubMed.
- K. Toyoda, R. Hinogami, N. Miyata and M. Aizawa, J. Phys. Chem. C, 2015, 119, 6495–6501 CAS.
- M. Yu, T. I. Draskovic and Y. Wu, Phys. Chem. Chem. Phys., 2014, 16, 5026–5033 RSC.
- J. Ahmed, C. K. Blakely, J. Prakash, S. R. Bruno, M. Yu, Y. Wu and V. V. Poltavets, J. Alloys Compd., 2014, 591, 275–279 CrossRef CAS.
- T. Ahmad, R. Phul, P. Alam, I. H. Lone, M. Shahazad, J. Ahmed, T. Ahamad and S. M. Alshehri, RSC Adv., 2017, 7, 27549–27557 RSC.
- J. Ahmed, V. V. Poltavets, J. Prakash, S. M. Alshehri and T. Ahamad, J. Alloys Compd., 2016, 688, 1157–1161 CrossRef CAS.
- R. Hinogami, K. Toyoda, M. Aizawa, S. Yoshii, T. Kawasaki and H. Gyoten, Electrochem. Commun., 2013, 35, 142–145 CrossRef CAS.
- J. Ahmed and Y. Mao, J. Solid State Chem., 2016, 242, 77–85 CrossRef CAS.
- C. G. Read, Y. Park and K. Choi, J. Phys. Chem. Lett., 2012, 3, 1872–1876 CrossRef CAS PubMed.
- T. I. Draskovic, M. Yu and Y. Wu, Inorg. Chem., 2015, 54, 5519–5526 CrossRef CAS PubMed.
- A. M. Fathi, S. A. M. Abdel-Hameed, F. H. Margha and N. A. A. Ghany, Z. Phys. Chem., 2016, 230, 1519–1530 CrossRef CAS.
- A. P. Amrute, Z. Łodziana, C. Mondelli, F. Krumeich and J. Pérez-Ramírez, Chem. Mater., 2013, 25, 4423–4435 CrossRef CAS.
- D. Xiong, Z. Xu, X. Zeng, W. Zhang, W. Chen, X. Xu, M. Wang and Y.-B. Cheng, J. Mater. Chem., 2012, 22, 24760–24768 RSC.
- D. Xiong, W. Zhang, X. Zeng, Z. Xu, W. Chen, J. Cui, M. Wang, L. Sun and Y.-B. Cheng, ChemSusChem, 2013, 6, 1432–1437 CrossRef CAS PubMed.
- S. Powar, D. Xiong, T. Daeneke, M. T. Ma, A. Gupta, G. Lee, S. Makuta, Y. Tachibana, W. Chen, L. Spiccia, Y.-B. Cheng, G. Götz, P. Bäuerle and U. Bach, J. Phys. Chem. C, 2014, 118, 16375–16379 CAS.
- D. Xiong, X. Zeng, W. Zhang, H. Wang, X. Zhao, W. Chen and Y.-B. Cheng, Inorg. Chem., 2014, 53, 4106–4116 CrossRef CAS PubMed.
- Z. Xu, D. Xiong, H. Wang, W. Zhang, X. Zeng, L. Ming, W. Chen, X. Xu, J. Cui, M. Wang, S. Powar, U. Bach and Y.-B. Cheng, J. Mater. Chem. A, 2014, 2, 2968–2976 CAS.
- D. Xiong, H. Wang, W. Zhang, X. Zeng, H. Chang, X. Zhao, W. Chen and Y.-B. Cheng, J. Alloys Compd., 2015, 642, 104–110 CrossRef CAS.
- D. Xiong, Y. Qi, X. Li, X. Liu, H. Tao, W. Chen and X. Zhao, RSC Adv., 2015, 5, 49280–49286 RSC.
- D. Xiong, Q. Zhang, Z. Du, S. K. Verma, H. Li and X. Zhao, New J. Chem., 2016, 40, 6498–6504 RSC.
- J.-P. Doumerc, A. Wichainchai, A. Ammar, M. Pouchard and P. Hagenmuller, Mater. Res. Bull., 1986, 21, 745–752 CrossRef CAS.
- M. Beekman, J. Salvador, X. Shi, G. S. Nolas and J. Yang, J. Alloys Compd., 2010, 489, 336–338 CrossRef CAS.
- W. C. Sheets, E. Mugnier, A. Barnabé, T. J. Marks and K. R. Poeppelmeier, Chem. Mater., 2006, 18, 7–20 CrossRef CAS.
- H. Hu, M. Zhu, F. Xie, N. Lei, J. Chen, Y. Hou and H. Yan, J. Am. Ceram. Soc., 2009, 92, 2039–2045 CrossRef CAS.
- W. Li, X. Gao, D. Xiong, F. Wei, W.-G. Song, J. Xu and L. Liu, Adv. Energy Mater., 2017, 7, 1602579 CrossRef.
- D. Xiong, Q. Zhang, S. M. Thalluri, J. Xu, W. Li, X. Fu and L. Liu, Chem. – Eur. J., 2017, 23, 8749–8755 CrossRef CAS PubMed.
- D. Xiong, W. Li and L. Liu, Chem. – Asian J., 2017, 12, 543–551 CrossRef CAS PubMed.
- M. Yu, G. Natu, Z. Ji and Y. Wu, J. Phys. Chem. Lett., 2012, 3, 1074–1078 CrossRef CAS PubMed.
- Y. Zhu, W. Zhou, J. Yu, Y. Chen, M. Liu and Z. Shao, Chem. Mater., 2016, 28, 1691–1697 CrossRef CAS.
- W. Zhou, M. Zhao, F. Liang, S. C. Smith and Z. Zhu, Mater. Horiz., 2015, 2, 495–501 RSC.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y.-L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS PubMed.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- S. Jung, C. C. McCrory, I. M. Ferrer, J. C. Peters and T. F. Jaramillo, J. Mater. Chem. A, 2016, 4, 3068–3076 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qi00621g |
| This journal is © the Partner Organisations 2018 |