
Synthesis, photophysical properties and cation-binding studies of bipyridine-functionalized gold(I) complexes†
Igor V.
Solovyev
 ab,
Aleksandar
Kondinski
c,
Kirill Yu.
Monakhov
c,
Igor O.
Koshevoy
*b and
Elena V.
Grachova
*a
ab,
Aleksandar
Kondinski
c,
Kirill Yu.
Monakhov
c,
Igor O.
Koshevoy
*b and
Elena V.
Grachova
*a
aInstitute of Chemistry, St. Petersburg State University, Universitetskii pr. 26, 198504 St. Petersburg, Russia. E-mail: e.grachova@spbu.ru
bDepartment of Chemistry, University of Eastern Finland, 80101 Joensuu, Finland. E-mail: igor.koshevoy@joensuu.fi
cInstitut für Anorganische Chemie, RWTH Aachen University, Landoltweg 1, 52074 Aachen, Germany
First published on 3rd November 2017
Abstract
Gold(I) has been used as an interlocking center to design five phenyl-bipyridine based complexes [LAu(L1)] and [RAu(L2)] where L1 = 5-(4-ethynylphenyl)-2,2′-bipyridine and L2 = 5-(4-(diphenylphosphino)phenyl)-2,2′-bipyridine (L = P(C6H11)3, 1; C6H3(CH3)2NC, 2; R = Ph2OHCC2, 3; PhC2, 4; C6F5, 5). The obtained compounds have been characterized by polynuclear NMR spectroscopy and X-ray crystallography. Investigation of their UV-Vis absorption and emission characteristics has been supported by density functional theory (DFT) calculations that rationalize the tunable electronic properties of these compounds. Complexation of the metal cations and the corresponding optical responses of [LAu(L1)], in particular for Cd(II) and Pb(II), indicate the important role of gold(I) ions in modulating the luminescence characteristics of these species upon binding the analyte cations.
Introduction
Interest in the rational design of tailor-made molecules that could be used as luminescent sensors for cation detection has significantly increased over the past few years.1–3 Detection of metal cations is a task of high importance since many of them are involved in biological processes (e.g. sodium, potassium, magnesium, calcium, lithium, zinc, iron, copper, etc.) and serve as enzyme cofactors (e.g. zinc, iron, and manganese), while some are very toxic for organisms and the environment (e.g. aluminum, mercury, lead, and especially cadmium). Fluorescent tools for tracing of e.g. cadmium are still underdeveloped4–7 and their further elaboration has to be pursued. The possibility of a direct detection of metal ions in solution following a luminescence response of a sensor allows for the significant reduction of the detection limit that is critically important for many applications including clinical diagnosis.To date, numerous luminescent sensors for cations have been developed,8 all of which rely on two molecular subunits, a receptor and a signaling moiety. The receptor subunit captures the cationic analyte, while the signaling one changes its luminescence properties (quenching, enhancement or color change of emitted light), thus providing a visible response that is facile to detect.1–3,9,10
Although a classical fluorescent sensor is usually built up of a pure organic π-conjugated system, phosphorescent sensors based on transition metal complexes are becoming more popular due to the attractive metal-directed luminescence characteristics, which are different from those of organic chromophores, such as large Stokes shift, tunable emission energies, high quantum yield and long emission lifetimes.10,11
Regardless of the nature of the signaling subunit, a limited set of pendant groups is usually used for the receptor subunit. It is represented by, for example, acyclic, macrocyclic, crown, calixarene, terpyridine, hydroxyquinoline or related ligand scaffolds that are capable of coordinating metal cations.1–3,9–11 Recently, a new fluorescent sensor decorated by a modular peptoid receptor unit was reported.12 In general, the design principles, which involve the choice of transition metal for the signaling subunit, typical approaches for linking of the receptor to the phosphorescent signaling subunit, coordination chemistry of metal probes, and energy transfer mechanisms within the sensor, have been discussed in detail.8,10,11
Gold(I) complexes, decorated by polytopic ligands with a pendant function that enables secondary coordination of a heteroion, play an important role in the modern chemistry of this noble metal.13 The unique electronic properties of closed-shell d10 Au(I) centers, which determine the rich photophysical behavior of organogold compounds, make these species particularly appealing candidates as molecular luminescent sensors for the detection of various metal ions.14–16
Today, luminescence sensing properties of gold(I) alkynyl complexes trigger much interest that is based upon the simple, linear, and rigid structure of these compounds, their stability and facile preparation, and a conceptually straightforward route to functionalize the ligand environment with a π-conjugated system, which is usually responsible for diverse optical properties.15,17 Polytopic ligands with bipyridine or terpyridine functions are traditionally popular in the design of this type of luminescent cation sensor or heterometallic molecular assembly.18–27 On the other hand, the phenomenon of aurophilicity associated with polynuclear gold(I) complexes provides the ability to switch photoluminescence properties as a result of the modulation of Au–Au interactions due to the change of the metal framework geometry upon binding of the analyte by the receptor unit.14 A few effective examples of aurophilicity14,28–31 and metallophilicity16,32 utilization for metal ion sensing based on a “host–guest” interaction at the molecular periphery of coordination complexes have been demonstrated. However, to the best of our knowledge, only two cases of mononuclear Au(I) compounds, one of which is represented by an elegant complex that demonstrates a luminescence response to both cations and anions, have been reported up to now.33,34
Herein we describe the synthetic design and photophysical properties of a family of mononuclear Au(I) alkynyl complexes decorated with a phenyl-bipyridine receptor subunit, which show attractive sensing ability toward lead(II) and cadmium(II) cations.
Experiment
General remarks, materials, and instruments
Ethynyltrimethylsilane, n-butyllithium (solution 1.6 M in hexane) and chlorodiphenylphosphine were commercially available and used without additional purification. Solvents were purified and distilled using standard procedures.35 5-(4-Bromophenyl)-2,2′-bipyridine was synthesized according to a published procedure.36 [(THT)AuCl]37 and [(C6F5)Au(THT)]38 precursor compound were prepared by a general method and [PhC2Au]n and [Ph2OHCC2Au]n were prepared according to reported procedures.39 Solution 1H, 1H1H COSY, 19F and 31P{1H} NMR spectra were recorded on a Bruker Avance 400 spectrometer. Elemental analyses were carried out with an Elementar Analysensysteme vario MICRO for 1, 3, and 4 and with a Eurovector Euro-EA3028-HT for L1, L2, and 2. Mass spectra were recorded on a Bruker Esquire 3000 PLUS with the electrospray (ESI) technique.Photophysical measurements in solution were carried out using 1,2-dichloroethane that was distilled prior to use. UV-Vis spectra were recorded on a Shimadzu UV-1800 spectrophotometer at a concentration of 10−5 M (1 cm cuvette). Emission spectra in solution were recorded on a Fluoromax (JY Horiba Inc.) spectrofluorimeter using a concentration of ca. 10−5 M. The absolute emission quantum yield in solution was determined by the comparative method using Coumarin-102 in ethanol as the reference with the refraction indexes of 1,2-dichloroethane and ethanol equal to 1.44 and 1.36 respectively.40 Luminescence lifetimes were determined by the Time-Correlated Single Photon Counting (TCSPC) method. The lifetime data were fitted using the Jobin–Yvon software package and the Origin 8.1 program.
Anal. calcd for C18H12N2: C, 84.35; H, 4.72; N, 10.93. Found: C, 84.30; H, 4.54; N, 10.73%. 1H NMR (CDCl3, 298 K): δ 8.94 (d, JHH 1.9 Hz, 1H), 8.73 (d, JHH 4.1 Hz, 1H), 8.52 (d, JHH 8.4 Hz, 1H), 8.47 (d, JHH 8.1 Hz, 1H), 8.04 (dd, JHH 8.3, 2.4 Hz, 1H), 7.87 (td, JHH 7.7, 1.7 Hz, 1H), 7.65 (s, 4H), 7.35 (ddd, JHH 7.4, 4.8, 1.0 Hz, 1H), 3.19 (s, 1H) ppm. FTIR (Nujol, cm−1): ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C) 2103w.
C) 2103w.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as the eluent. Yield: 290 mg (44%).
:1) as the eluent. Yield: 290 mg (44%).
Anal. calcd for C28H21N2P: C, 80.75; H, 5.08; N, 6.73. Found: C, 80.92; H, 5.09; N, 6.31%. 1H NMR (CDCl3, 298 K): δ 8.95 (d, JHH 2.0 Hz, 1H), 8.73 (d, JHH 4.5 Hz, 1H), 8.50 (d, JHH 8.3 Hz, 1H), 8.46 (d, JHH 7.9 Hz, 1H), 8.05 (dd, JHH 8.1, 2.4 Hz, 1H), 7.86 (td, JHH 7.6, 1.8 Hz, 1H), 7.66 (d, JHH 8.1 Hz, 2H), 7.46 (t, JHH 7.4 Hz, 2H), 7.40 and 7.39 (s, 10H), 3.19 (c, 1H), 7.35 (ddd, JHH 7.5, 4.8, 1.0 Hz, 1H) ppm. 31P{1H} NMR (CDCl3; 298 K): δ −5.8 (s, 1P) ppm.
C) 2109w.
C) 2115m, ν(NC) 2208m.
X-ray structure determination
Single crystals of 1, 2, 5 and 2·Pb were grown by slow gas phase diffusion of pentane into CH2Cl2 (1, 2, 5) or acetonitrile/C2H4Cl2 (2·Pb) solutions at ambient temperature. The crystals were immersed in cryo-oil, mounted in a nylon loop, and measured at a temperature of 120 K (1), 150 K (2, 5) or 100 K (2·Pb). The X-ray diffraction data were collected with Bruker Smart Apex, Kappa Apex II Duo and Agilent Technologies SuperNova Atlas diffractometers using MoKα (λ = 0.71073 Å) or CuKα (λ = 1.54184 Å) radiation. The APEX241 and CrysAlis Pro42 program packages were used for cell refinements and data reductions. The structures were solved by direct methods using the SHELXS-2014 programs43 with the WinGX graphical user interface.44 A semi-empirical absorption correction (SADABS45 or CrysAlis Pro42) was applied to all data. Structural refinements were carried out using SHELXL-2014.43 The crystallization solvent in 1 and 2·Pb was partially lost and could not be resolved unambiguously. Its contribution to the calculated structure factors was taken into account by using a SQUEEZE routine of PLATON.46 The missing solvent was not taken into account in the unit cell content. The structure of 2 was refined as a 2-component inversion twin. All hydrogen atoms were positioned geometrically and constrained to ride on their parent atoms, with C–H = 0.95–0.98 Å, and Uiso = 1.2 − 1.5Ueq (parent atom). The crystallographic details are summarized in Table S1.†Computational methods
The DFT and TD-DFT calculations were performed with the Amsterdam Density Functional code (ADF 2016).47,48 Basis sets of triple-zeta plus one polarization quality (TZP)49 were used for all atoms. Scalar relativistic corrections were taken into account within the zero-order regular approximation (ZORA),50,51 and the solvent was modeled using the conductor-like screening model (COSMO)52–54 with parameters for dichloroethane (ε = 10.66; solvent radius = 3.15 Å). DFT geometry optimizations were performed using the BP86 functional,55,56i.e. at the BP86/ZORA-scalar/TZP/COSMO level of theory. The B3LYP functional57 together with triple-zeta singly polarized basis sets (i.e., B3LYP/ZORA-scalar/TZP/COSMO) was used to carry out the TD-DFT calculations of the full-electronic spectra for all studied structures. The lowest 20 allowed excitations were calculated (with the Davidson algorithm) and used to simulate the full absorption spectra. This number of excitations was sufficient to define the entire spectrum down to 250 nm. The theoretical spectra (shown in Fig. SC4†) were obtained by convolution of Gaussian curves using a convolution factor (peak width) of 50 nm. All energies discussed herein are based on the calculations at B3LYP/ZORA-scalar/TZP/COSMO.Results and discussion
Synthesis and NMR characterization of gold(I) complexes
The ditopic ligands L1 and L2, synthesized as shown in Scheme 1, are light-yellow crystalline solids, stable to moisture and air, and readily soluble in common polar organic solvents. The structure and integrity of both compounds in solution were evidenced by their 1H NMR spectroscopic characteristics (number of resonances, chemical shifts, relative intensities and multiplicity; see Experimental and Fig. S1 and S2†). Expectedly, the proton spectra of the bipyridine part of L1 and L2 are nearly identical; a difference was observed only for the {C6H4} fragment. | ||
| Scheme 1 Synthesis of ligands L1 and L2. | ||
Compounds L1 and L2 were used to synthesize mononuclear Au(I) complexes 1–5. The synthetic route includes (a) depolymerization of a gold acetylenide by a neutral co-ligand like phosphine or isocyanide in a dichloromethane solution (for 1–4), or (b) interaction of a labile gold complex with phosphine (for 5),32,39,58–61 as illustrated in Schemes 2 and 3.
 | ||
| Scheme 2 Synthesis of complexes 1 and 2. | ||
 | ||
| Scheme 3 Synthesis of complexes 3–5. | ||
Compounds 1–5 were isolated as pale yellow or colorless solids and their composition was confirmed by CHN analysis. All Au(I) complexes obtained were characterized in solution by polynuclear NMR spectroscopy. 31P NMR spectra of 1 and 3–5 exhibit one singlet resonance at 56.3, 41.7, 42.0, and 36.5 ppm, respectively, which demonstrate a low field shift, typical for the Au-coordinated phosphine ligands. Assignment of proton signals for 1–5 (Fig. S1–S3†) was performed on the basis of 1H1H COSY spectra (Fig. S5–S8†). A comparison of 1H NMR spectra of L1 and L2 to those of their corresponding complexes shows that coordination of the metal did not influence the spectral characteristics of the bipyridine part and affected only the signals of the {C6H4} fragment in 1–5 as well as those of the phenyl rings at phosphorus in 3–5 (Fig. S1–S3†). The 19F NMR spectrum of 5 consists of tree multiplets as expected (Fig. S4†).
Complexation with heterometal cations
Addition of Cd(II) or Pb(II) salts to solution of complexes 1 or 2 results in significant changes in the 1H NMR spectra (Fig. 1 and S9–S11†). The resonances of the protons H1–H6 and H11 of the bipyridyl part shift toward low field, which provides a support for coordination of the heterometal cations to the free bipyridine function of 1 or 2. | ||
| Fig. 1 1H NMR spectrum of 1 + Pb(ClO4)2; CDCl3 + CD3CN, r.t., aromatic range; signals of CDCl3 residual protons are marked by asterisks. | ||
1H NMR spectra clearly indicate that the interaction of the Au(I) complexes 1 and 2 with heterometal cations leads to the formation of an adduct “complex:M(II)”. We assume that the broadening of the signals is a result of the “dissociation/association” equilibrium between the adduct, native complex, and heterometal cation at room temperature.
Structural characterization
Molecular structures of complexes 1, 2, and 5 (Fig. 2) were established by X-ray diffraction (XRD) analysis and match the NMR spectroscopic data (see Schemes 2 and 3). Selected structural parameters are given in Table 1. The crystal structures of 1, 2, and 5 feature the expected binding of ligands L1 and L2 to the gold(I) centers and a linear coordination geometry of the metal ions that is typical for Au(I) complexes. The bipyridine part of L1 and L2 in the studied structures is not involved in any coordination as indicated by the NMR experiments. The bond lengths around the metal center fall in the range typical for other mono-18,20,22,24,28,33,62,63 and polynuclear59,64–68 Au(I) compounds that contain {P–Au–CC}, {NC–Au–CC} or {P–Au–C6F5} moieties (Table 1). These fragments in 1, 2, and 5 adopt an almost ideal linear geometry that is accompanied by nearly eclipsed conformation of all three phenyl-bipyridine aromatic rings of L1 and L2. Analysis of the crystal packing reveals no aurophilic interactions in the structures of 1, 2, and 5, while some intermolecular, relatively short contacts between protons and the aromatic system of the phenyl-bipyridine part in 1 and 2 as well as between fluorine and the protons of the phenyl ring in 5 are observed. These weak interactions however do not result in substantial distortions of the molecular structure.
 | ||
| Fig. 2 Molecular structures of complexes 1, 2, and 5. | ||
| Bond lengths, Å | Angles, ° | ||
|---|---|---|---|
| 1 | |||
| P1–Au1 | 2.2990(6) | P1–Au1–C1 | 175.42(6) |
| Au1–C1 | 2.008(2) | Au1–C1–C2 | 177.5(2) |
| C1–C2 | 1.208(3) | C1–C2–C3 | 178.2(2) |
| 2 | |||
| N3–C19 | 1.13(2) | N3–C19–Au1 | 180(1) |
| C19–Au1 | 1.97(1) | C19–Au1–C1 | 177.8(5) |
| Au1–C1 | 1.95(1) | Au1–C1–C2 | 174(1) |
| C1–C2 | 1.22(2) | C1–C2–C3 | 177(1) |
| 5 | |||
| Au1–P1 | 2.2642(8) | P1–Au1–C1 | 176.63(8) |
| Au1–C1 | 2.047(2) | ||
Thus, the NMR spectroscopic and XRD data suggest that the bipyridyl function in complexes 1–5 can be exploited toward complexation with heterometals to form molecular adducts. Indeed, NMR spectra indicated that in solution complexes 1 and 2 effectively bind metal cations such as Pb(II) and Cd(II). Single crystals of the anticipated 2·Pb adduct with a ratio of 2:Pb(ClO4)2 = 1:1 were isolated. Unfortunately, the inherent tendency of 2·Pb for easy dissociation in solution did not allow selective isolation as crystallization attempts produced inseparable mixtures of 2·Pb and of the starting compounds. The molecular structure of 2·Pb is illustrated in Fig. 3 and selected structural parameters are given in Table 2.
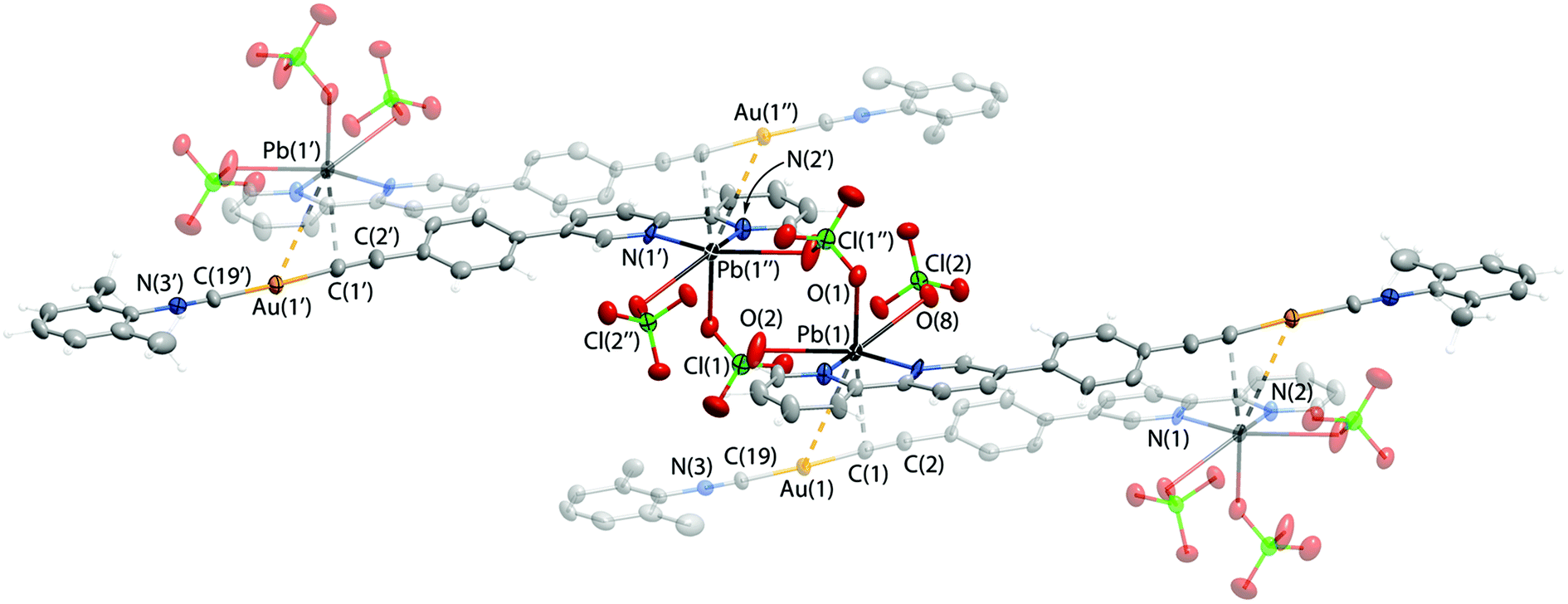 | ||
| Fig. 3 Molecular structure of 2·Pb adduct. | ||
| Bond lengths, Å | Angles, ° | ||
| N3–C19 | 1.13(1) | N3–C19–Au1 | 178(1) |
| C19–Au1 | 1.98(1) | C19–Au1–C1 | 177.0(5) |
| Au1–C1 | 1.97(1) | Au1–C1–C2 | 177(1) |
| C1–C2 | 1.24(2) | C1–C2–C3 | 172(1) |
| Pb1–N2 | 2.478(7) | Contacts, Å | |
| Pb1–N1 | 2.439(8) | Pb1⋯Au1 | 3.5810(6) |
| Pb1–O8 | 2.776(7) | Pb1⋯C1 | 2.85(1) |
| Pb1–O1 | 2.708(8) | ||
| Pb1–O2 | 2.819(9) | ||
Coordination of Pb(II) to the bipyridine part of 2 affords a chelate cycle, and analysis of the CSD allows the conclusion that there are no anomalies in the structure of this 5-membered “C2N2Pb” ring. Adduct formation does not influence the Au(I) segment of 2·Pb, which remains unchanged after Pb(II) coordination (Table 2).
In the crystal structure, molecules of 2·Pb were found to form a supramolecular polymer via two bridging ClO4− anions that coordinate to two Pb(II) ions. Although ClO4− acts as a ligand for Pb(II) quite often, there are only two studies that describe a κ2,μ2-ClO4− binding mode featuring short Pb–O bond lengths similar to those observed for 2·Pb.69,70 Also, the crystal structure of 2·Pb displays an extremely rare unsupported Au(I)⋯Pb(II) interaction with a distance of 3.5810(6) Å between these two atoms (Fig. 3). To date, there are only two structurally characterized species that exhibit metallophilic interactions with Au⋯Pb contacts of 3.0494(4) Å and 3.7115(1) Å.71,72
Thus, three ClO4− anions attached to the Pb(II) ion complement the coordination environment of the heterometal ion and together with {Au(I)⋯Pb(II)⋯C} interactions adopt the distorted octahedral geometry of the Pb(II) ligand sphere. On the other hand, Au⋯Pb bonding as well as additional weak interactions between the Pb(II) ion and the alkynyl carbon atom of a neighboring molecule give rise to a supramolecular chain in the solid state of 2·Pb.
Optical and photophysical properties of 1–5
The absorption spectra of ligands L1 and L2 and complexes 1–5 are substantially comparable due to the similarity of the chromophoric centers localized on the extended aromatic system (Fig. 4 and Table 3). The most intense absorption band about 310 nm in the spectra of L1 and L2 and of complexes 3–5 corresponds to intraligand π → π* transitions localized at a conjugated aromatic system of the phenyl-bipyridine segment that is common for all compounds studied. Evidently, due to electronic communication between the chromophoric center and Au(I) through the CC bond this band undergoes a red-shift in the spectra of complexes 1 and 2. This shift allows the extension of the excitation wavelength range for 1 and 2, thereby increasing the importance of these compounds as potential sensors. The high-energy absorption bands in the region of 250–275 nm correspond to intraligand π → π* transitions in aromatic/phenyl rings of dimethylphenyl isocyanide (2), phosphine (3–5), and alkynyl ligands (3, 4). The low intensity band about 375 nm in the spectra of 1 and 2 can presumably be originated from 1MLCT transitions (vide infra).
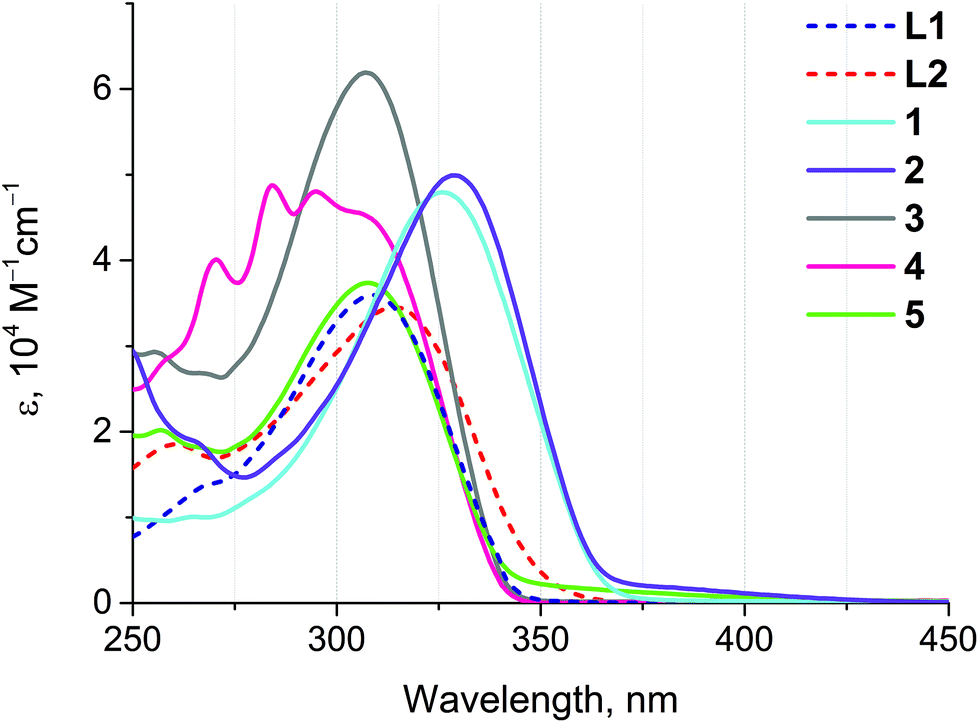 | ||
| Fig. 4 UV-Vis absorption spectra of ligands L1 and L2 and complexes 1–5 measured in dichloroethane at r.t. | ||
| λ abs/nm (ε 104/M−1 cm−1) | λ em/nm (λexcit/nm) | τ/ns | |
|---|---|---|---|
| a Vibronic structure with ∼1300 cm−1 spacing. Emission spectrum of L1 is given in Fig. S12. | |||
| L1 | 308 (3.6) | 367a (310) | 0.50 |
| L2 | 313 (3.5), 260 (1.8) | — | — |
| 1 | 326 (4.8), 265 (1.0) | 387 (335) | 0.49 |
| 2 | 329 (4.9), 266 (1.9) | 383 (330) | 1.52 |
| 3 | 307 (6.2), 268 (2.7), 255 (2.9) | — | — |
| 4 | 307 (4.5), 295 (4.8), 284 (4.9), 270 (4.0), 257 (2.8) | — | — |
| 5 | 308 (3.7), 257 (2.0) | — | — |
Interestingly, complexes 3–5 are not emissive in solution and in the solid state, while 1 and 2 show weak emission in solution at ambient temperature with quantum yields of ∼1% (see Fig. 5 and Table 3). We speculate that the emission of complexes is low up to total blanking possibly because of quenching by photoinduced electron transfer (PET) from the N lone pairs in the bipyridine part.3,8,10,73 The photophysical properties of the congener phenyl-bipyridines were evaluated previously36 and revealed very strong dependence of quantum efficiency on the electronic properties of the substituents in the 5-position of the bipy motif. The presence of moderately electron-deficient groups (including phenyl) resulted in low quantum yields (Φ = 0.8% for {4-Cl-C6H4} group) that were assigned to the contribution of nitrogen lone pairs to the excited states (n → π* transitions) and the appearance of non-radiative decay pathways. Therefore, it is not surprising that introducing more electron accepting groups (alkynyl in 1 and 2 and quaternized phosphine motif in 3–5) causes poor emission for these gold complexes.
 | ||
| Fig. 5 Normalized excitation (dashed line) and emission (solid line) spectra of 1 and 2 obtained in dichloroethane solution at r.t. The excitation wavelengths are indicated on the diagram. | ||
1 and 2 display shortwave emission maximized at about 380 nm with a small Stokes shift and lifetime in the nanosecond domain, which may be assigned to intraligand fluorescence localized on the phenyl-bipyridyl moieties slightly perturbed by coordination to Au(I), namely to the 1IL(π → π*) transition; the absence of oxygen quenching of emission confirms this conclusion. Fluorescence of gold(I) complexes with extended aromatic systems is not a rare phenomenon. The ratio of singlet and triplet emission intensity in this type of complex depends on many parameters including the spatial separation between the metal and ππ* chromophoric center. The relatively long distance between Au(I) and the emissive bipyridine fragment in 1 and 2 results in a weak spin–orbit coupling (SOC) and consequently in retardation of intersystem crossing (ISC) that leads to the dominating fluorescence emission in these complexes.62,74–78
Photophysical response of 1 and 2 to metal cations in solution
Complexes 1 and 2 show a distinct alteration of their photophysical properties in the presence of metal ions. The main band in the absorption spectra decreases with an increase in the concentration of the heterometal cation. Intensities of the new absorption bands with shorter and longer wavelengths increase simultaneously (Fig. 6, example of 1 + Pb(ClO4)2). These changes are significant up to the molar ratio of complex:heterometal = 2:1 and become less pronounced with further growth of the concentration of the Pb(II) salt. Analogous behavior has been observed for all systems studied herein (Fig. S13†). Since the main band in the absorption spectra of 1 and 2 refers to the intraligand π → π* transitions localized at a conjugated aromatic system of phenyl-bipyridine, the observed changes should be related to coordination of the heterometal to the diimine fragment. The increase of the low energy band at 363 nm upon addition of M(II) can be attributed to the ligand-to-metal charge transfer (LMCT) arising from the transfer of electron density from the lone pairs of the bipyridyl moiety to M(II), vide infra.
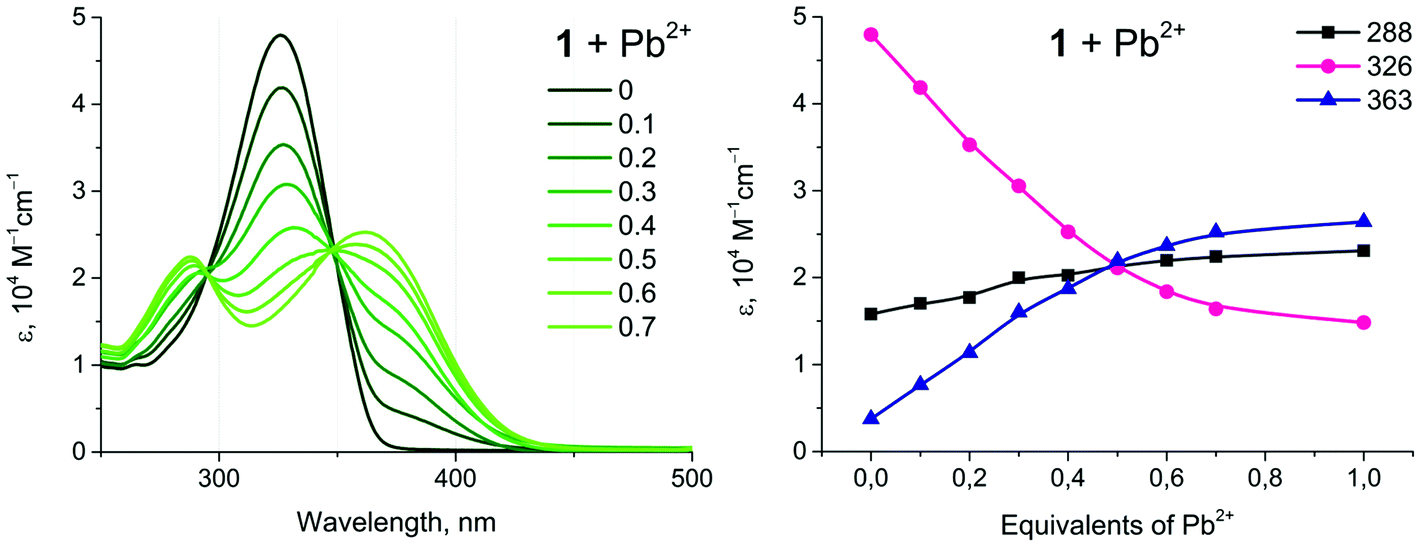 | ||
| Fig. 6 Left: UV-Vis absorption spectra of 1 (10−5 M) under Pb(ClO4)2 titration; molar equivalents of Pb(II) are indicated on diagram, dichloroethane, r.t. Right: The dependence of the extinction coefficient at different wavelengths on molar equivalents of Pb(II). | ||
To showcase the selectivity and sensitivity of heterometal cation detection we studied the luminescence response of 1 and 2 for several metal cations, namely Cd(II), Pb(II), K(I), Co(II), Cu(II), Cr(III), Hg(II), and Na(I). It was observed that complexes 1 and 2 show different emission changes upon binding metal ions. The presence of Co(II) or Cu(II) in solution decreases the fluorescence intensity of 2 down to complete quenching. An analogous phenomenon has been reported previously for mononuclear Au(I) complexes functionalized with a dipicolylamine receptor.33 For the other heterometals 1 demonstrated approximately the same changes in fluorescence intensity (Fig. S14†), whereas the fluorescence of 2 was enhanced by 3 times in the presence of Pb(II) and by 25 times in the presence of Cd(II) (see Fig. 7). This dramatic increase of the emission efficiency can be attributed to coordination of the bipy function to the heterometals that excludes the detrimental effect of nitrogen lone pairs and blocks the possibility of non-radiative relaxation.
 | ||
| Fig. 7 Left: Ratio of integral emission intensity Iadduct/I2, measured in dichloroethane solution at r.t. Right: Solution of 2 with a 10−5 M concentration and the same solution after addition of a ∼0.1 molar equivalent of Cd(ClO4)2, under a UV lamp (365 nm). | ||
Titration of 2 with M(ClO4)2 results in a ratiometric growth of the red-shifted emission band upon excitation at 330 nm. The emission band of the native Au(I) complex disappears completely at the ratio of complex:heterometal = 2:1. The change in the emission intensity at 522 and 463 nm for Pb(II) and Cd(II), respectively, is a linear function of the M(II) concentration until a 0.5 molar equivalent of heterometal cation (Fig. 8). Quantitative characteristics of the adduct fluorescence properties are summarized in Table S2.† It is noteworthy that a further increase in the amount of heterometal leads to the realization of dynamic equilibrium between several forms of adducts with different ratios Au : heterometal. This is in good agreement with the proton spectra of the system (see Fig. 1 and S9–S11†) and with the loss of ratiometric dependence of luminescence response from heterometal concentration.
 | ||
| Fig. 8 Left: Emission spectra of 2 (10−5 M) upon treatment with M(ClO4)2, measured in dichloroethane at r.t. Right: Plots of emission integral intensity as a function of the M(II) concentration. | ||
Furthermore, it has to be mentioned that the interaction of Pb(II)/Cd(II) with L1 also results in visible changes of the ligand luminescence properties. However, a much smaller red shift and only a slight increase in the fluorescence of L1 have been observed (Fig. S15†). Thus, the use of a gold(I) complex as a sensor is justified from the viewpoint of convenient luminescence excitation and easy measurement of its variations. In particular, the importance of Au(I) ions is highlighted by the substantial decrease of emission energy of 2:Pb vs.2, which can be tentatively ascribed to the formation of the unconventional Au⋯Pb interaction.
Typically, two types of electronic transition, namely ligand centered (LC) and ligand-to-ligand charge transfer (LLCT), seem to be responsible for the emission of d10 closed-shell Cd(II) coordination complexes with diimine ligands.79,80 On the other hand, involving the low lying s or p empty orbitals of Cd(II) leads to the observation of LMCT states. Moreover, usually the ligand provides σ* antibonding orbitals to the LUMO, and the lowest excited level is more accurately described as a mixture of LMCT and LC/LLCT states.81
DFT calculations
The synthesized compounds and their precursors were fully optimized in solution as discrete species (see Fig. SC1†). The calculated geometry parameters (bond lengths and bond angles) of native complexes 1, 2 and 5 have been shown to be in good agreement with those determined experimentally (see Table SC1†). DFT calculations have revealed that ligand L1 exhibits the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) resembling a typical π and π* system, that are well delocalized (see Fig. 9). L1 exhibits a HOMO–LUMO energy gap of 4.1 eV. In contrast to L1, complexes 1 and 2 exhibit lower gaps of ca. 3.8 eV. Such a reduction in the HOMO–LUMO energy gap can be related to the higher lying HOMO energies of 1 and 2 due to the antibonding interactions between Au-centered d atomic-like orbitals and the π-like orbitals situated on the neighboring {CC} moiety. The LUMO of 1 exhibits a π* system that extends over the phenylene-bipyridine segment, resembling that of L1. The LUMO of 2 extends over the whole C/N framework including the segment of the dimethylphenyl isocyanide moiety. The LUMO of 2 also exhibits small contributions of Au-centered d atomic-like orbitals.
 | ||
| Fig. 9 Frontier molecular orbitals involved in the first excitation of L1, 1 and 2. Color code: Au = yellow, P = pink, C = gray, N = blue, and H = white. | ||
In contrast to L1, the HOMO of L2 exhibits an electron lone pair positioned at the phosphorus center as well as numerous contributions of localized π bonding orbitals (Fig. SC2†). The LUMO of L2 is delocalized over the phenylene-bipyridine segment and reveals a typical π* antibonding system. L2 exhibits a HOMO–LUMO energy gap of 4.0 eV which is similar to that of L1. Complexes 3 and 5 show a π bonding HOMO that is distributed primarily over the phenylene-bipyridine segment. In 4 HOMO−1 is qualitatively similar to the HOMOs of 3 and 5, while the HOMO is localized over the {Au–C2Ph} moiety and features Au-centered d atomic-like orbitals and π bonding orbitals over the alkynyl-phenyl system (Fig. SC2†). Both these frontier MOs are involved in the first excitation and discussed below (vide infra). The LUMOs of 3–5 are qualitatively and energetically similar featuring π* antibonding orbitals localized over the phenylene-bipyridine segment.
To understand how the electronic properties of these molecules relate to the UV-Vis spectroscopic properties, we performed TD-DFT calculations at the B3LYP level. The obtained results show that the calculated UV-Vis spectra follow the observed experimental trends and therefore allow a qualitative assignment of the major absorptions (see ESI† for more details). Compounds L1, L2, 1–3 and 5, with the exception of 4, exhibit first UV-Vis excitation that is predominantly defined by the HOMO → LUMO transition. In 4 both HOMO → LUMO and HOMO−1 → LUMO transitions contribute to the first excitation. Complexes 1 and 2 are characterized by first excitation energies of ca. 3.3 eV, which is lower than that of L1 with ca. 3.5 eV. Complexes 3–5 have first excitation of ca. 3.7 eV, which is larger in energy than that of L2 with ca. 3.5 eV. The transitions between the frontier MOs are relevant for the observation of luminescence in these compounds. While the first excitation in 3–5 relates to only π and π* excitation, the first excitation in 1 and 2 corresponds to π → π* transitions and Au(d) → π* metal to ligand charge transfer (MLCT) transitions. The MLCT allows depopulation of the Au(I) center and destabilization of the {CC} functionality. As this process is accompanied by separation of charges and by changes in geometries (e.g. extension in the C–C distance of the {CC} moiety), we assume that it represents a viable origin of the observed fluorescence in 1 and 2.
To shed light on the binding of Cd(II) centers to native 2 and its influences on the UV-Vis spectra we designed a model system abbreviated as 2·Cd(ClO4)2 (see Fig. 10). In this model system, complex 2 and two “bidentate” ClO4− anions bind to a single Cd(II) center, leading to calculated Cd–O distances of 2.395–2.500 Å. The latter fall within the expected range of Cd–O bond lengths when considering the related [Cd(bpy)2(H2O)(ClO4)]+ complex.82 Therefore, the 2·Cd(ClO4)2 model depicts a scenario with 2:Cd(II) of 1:1, while retaining neutrality and six-fold coordination of the Cd(II) center, typically observed for complexes of Cd(II) with bipyridine.83,84 The density isosurface and the molecular electrostatic potential (MEP) of 2·Cd(ClO4)2 point out the ionic interaction between the chlorate moieties and the Cd(II) center. Highly negative potentials on 2 appear in the vicinity of the {CC} bond. In comparison to 2, this model also shows red-shifted first excitation, calculated at ca. 4.0 eV, which is in line with experimental observations.
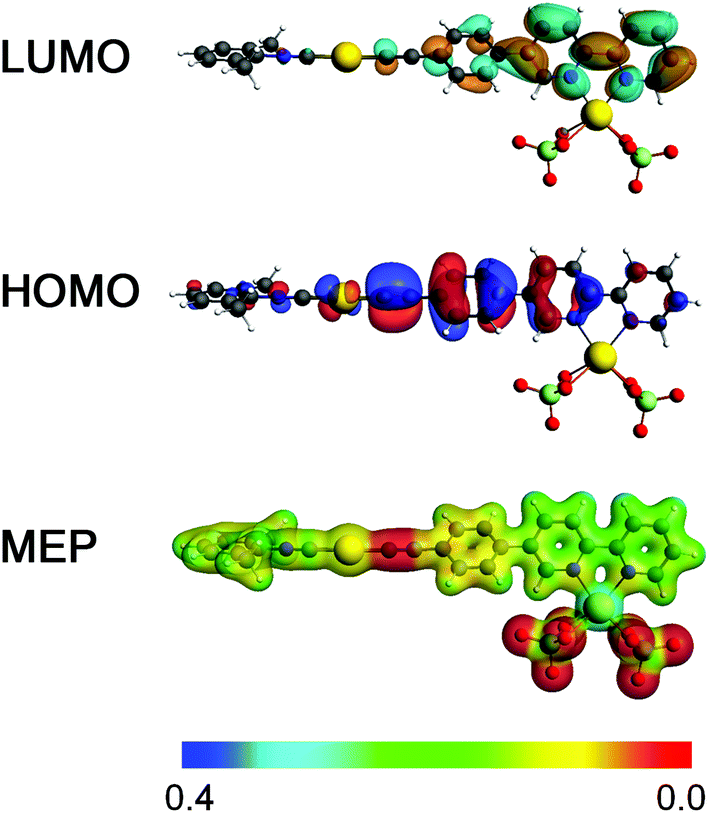 | ||
| Fig. 10 Depiction of frontier molecular orbitals and MEP of the 2·Cd(ClO4)2 adduct calculated at the B3LYP/ZORA-scalar/TZP/COSMO level of theory. The red regions feature the highest negative potentials. | ||
The first excitation in 2·Cd(ClO4)2 is dominated by the HOMO → LUMO transition. The HOMO of 2·Cd(ClO4)2 is characterized by Au-centered d atomic-like orbitals and π bonding orbitals over the {CC} moiety and the phenylene functionality attached to the bipyridine moiety. The LUMO in 2·Cd(ClO4)2 is represented by π* antibonding orbitals that are majorly localized over the bipyridine moiety as a result of Cd(II) binding. The direct HOMO → LUMO transition can be described as π → π* and Au(d) → π* transitions, and therefore this model adduct can be viewed as an intramolecular donor–acceptor system.
Conclusion
We presented the synthesis and spectroscopic characterization of a new family of gold(I) compounds that can serve as effective metalloligands due to a structurally exposed diimine functionality accessible for subsequent metal coordination. The parent complexes [LAu(L1)] and [RAu(L2)], where L1 is (ethynylphenyl)-bipyridine and L2 is ((diphenylphosphino)phenyl)-bipyridine, demonstrate poor to negligible fluorescence in solution, which is dramatically enhanced upon binding the metal cations and reaches maximum quantum yields of 11% and 44% for Pb(II) and Cd(II) ions, respectively. In addition to this ‘turn on’ response, the 2:Pb(II) adduct (2 = [LAu(L1)], L = 2,6-dimethylphenyl isocyanide) exhibits ca. 140 nm bathochromic shift of emission maximum with respect to the native complex 2. The X-ray crystallographic study of heterobimetallic compound 2·Pb provides support for the adduct formation through heterometal coordination to the bipyridine function of the diimine ligand. The clearly detectable photophysical changes of the title diimine-functionalized gold(I) complexes in the presence of selected metal ions make them promising candidates for chemosensing.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors greatly appreciate financial support from the Russian Science Foundation, grant 16-13-10064. The work was carried out using the equipment of core facilities of St Petersburg State University Research Park: Centers of Magnetic Resonance, Optical and Laser Materials Research, Chemical Analysis and Materials Research, and the X-ray Diffraction Centre. DFT calculations were performed using supercomputing facilities at the Institute of Inorganic Chemistry at RWTH Aachen University.References
- M. H. Keefe, K. D. Benkstein and J. T. Hupp, Coord. Chem. Rev., 2000, 205, 201–228 CrossRef CAS.
- M. A. Palacios, Z. Wang, V. A. Montes, G. V. Zyryanov and P. Anzenbacher, J. Am. Chem. Soc., 2008, 130, 10307–10314 CrossRef CAS PubMed.
- K. P. Carter, A. M. Young and A. E. Palmer, Chem. Rev., 2014, 114, 4564–4601 CrossRef CAS PubMed.
- G. Hennrich, S. Helmut and R.-G. Ute, J. Am. Chem. Soc., 1999, 121, 5073–5074 CrossRef CAS.
- H. N. Kim, W. X. Ren, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2012, 41, 3210–3244 RSC.
- X. Zhou, P. Li, Z. Shi, X. Tang, C. Chen and W. Liu, Inorg. Chem., 2012, 51, 9226–9231 CrossRef CAS PubMed.
- M. Taki, in Cadmium: From Toxicity to Essentiality, ed. A. Sigel, H. Sigel and R. K. O. Sigel, Springer Science + Business Media, Dordrecht, 2013, vol. 11, pp. 99–115 Search PubMed.
- M. C.-L. Yeung and V. W.-W. Yam, Chem. Soc. Rev., 2015, 44, 4192–4202 RSC.
- B. Valeur, Coord. Chem. Rev., 2000, 205, 3–40 CrossRef CAS.
- Z. Liu, W. He and Z. Guo, Chem. Soc. Rev., 2013, 42, 1568–1600 RSC.
- Q. Zhao, F. Li and C. Huang, Chem. Soc. Rev., 2010, 39, 3007–3030 RSC.
- A. S. Knight, R. U. Kulkarni, E. Y. Zhou, J. M. Franke, E. W. Miller and M. B. Francis, Chem. Commun., 2017, 53, 3477–3480 RSC.
- J. Gil-Rubio and J. Vicente, Chem. – Eur. J., 2017 DOI:10.1002/chem.201703574.
- X. He and V. W.-W. Yam, Coord. Chem. Rev., 2011, 255, 2111–2123 CrossRef CAS.
- J. Carlos Lima and L. Rodríguez, Chem. Soc. Rev., 2011, 40, 5442–5456 RSC.
- Y. Zhou, E. Liu, J. Wang and H. Chao, Inorg. Chem., 2013, 3, 8629–8637 CrossRef PubMed.
- V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed.
- N. C. Habermehl, F. Mohr, D. J. Eisler, M. C. Jennings and R. J. Puddephatt, Can. J. Chem., 2006, 84, 111–123 CrossRef CAS.
- R. Packheiser, P. Ecorchard, T. Ruffer, B. Walfort and H. Lang, Eur. J. Inorg. Chem., 2008, 2008, 4152–4165 CrossRef.
- R. Packheiser, A. Jakob, P. Ecorchard, B. Walfort and H. Lang, Organometallics, 2008, 27, 1214–1226 CrossRef CAS.
- H.-B. Xu, L.-Y. Zhang, J. Ni, H.-Y. Chao and Z.-N. Chen, Inorg. Chem., 2008, 47, 10744–10752 CrossRef CAS PubMed.
- X.-L. Li, K.-J. Zhang, J.-J. Li, X.-X. Cheng and Z.-N. Chen, Eur. J. Inorg. Chem., 2010, 2010, 3449–3457 CrossRef.
- J. Vicente, J. Gil-Rubio, N. Barquero, V. Camara and N. Masciocchi, Chem. Commun., 2010, 46, 1053–1055 RSC.
- X.-L. Li, M. Tan, K.-J. Zhang, B. Yang, J. Chen and Y.-B. Ai, Inorg. Chem., 2012, 51, 109–118 CrossRef CAS PubMed.
- V. Camara, N. Masciocchi, J. Gil-Rubio and J. Vicente, Chem. – Eur. J., 2014, 20, 1389–1402 CrossRef CAS PubMed.
- V. Camara, N. Barquero, D. Bautista, J. Gil-Rubio and J. Vicente, Chem. – Eur. J., 2015, 21, 1992–2002 CrossRef CAS PubMed.
- V. Camara, N. Barquero, D. Bautista, J. Gil-Rubio and J. Vicente, Inorg. Chem., 2015, 54, 6147–6156 CrossRef CAS PubMed.
- X. He, E. C.-C. Cheng, N. Zhu and V. W.-W. Yam, Chem. Commun., 2009, 27, 4016–4018 RSC.
- X. He, W. H. Lam, N. Zhu and V. W.-W. Yam, Chem. – Eur. J., 2009, 15, 8842–8851 CrossRef CAS PubMed.
- F. K.-W. Hau, X. He, W. H. Lam and V. W.-W. Yam, Chem. Commun., 2011, 47, 8778–8780 RSC.
- F. K.-W. Hau and V. W.-W. Yam, Dalton Trans., 2016, 45, 300–306 RSC.
- J. R. Shakirova, E. V. Grachova, V. V. Gurzhiy, I. O. Koshevoy, A. S. Melnikov, O. V. Sizova, S. P. Tunik and A. Laguna, Dalton Trans., 2012, 41, 2941–2949 RSC.
- X. He, N. Zhu and V. W.-W. Yam, Organometallics, 2009, 28, 3621–3624 CrossRef CAS.
- X. He and V. W.-W. Yam, Inorg. Chem., 2010, 49, 2273–2279 CrossRef CAS PubMed.
- W. L. F. Armarego and C. L. L. Chai, Purification of Organic Chemicals, 2009 Search PubMed.
- V. N. Kozhevnikov, O. V. Shabunina, D. S. Kopchuk, M. M. Ustinova, B. König and D. N. Kozhevnikov, Tetrahedron, 2008, 64, 8963–8973 CrossRef CAS.
- R. Uson, A. Laguna, M. Laguna, D. A. Briggs, H. H. Murray and J. P. Fackler, in Inorganic Syntheses, 2007, vol. 29, pp. 85–91 Search PubMed.
- R. Uson, A. Laguna and J. Vicente, J. Organomet. Chem., 1977, 131, 471–475 CrossRef CAS.
- I. O. Koshevoy, Y. C. Chang, A. J. Karttunen, S. I. Selivanov, J. Jänis, M. Haukka, T. Pakkanen, S. P. Tunik and P. T. Chou, Inorg. Chem., 2012, 51, 7392–7403 CrossRef CAS PubMed.
- K. Rurack and M. Spieles, Anal. Chem., 2011, 83, 1232–1242 CrossRef CAS PubMed.
- APEX2 Software Suite for Crystallographic Programs, Bruker AXS, Madison, Wisconsin, USA, 2010 Search PubMed.
- CrysAlis Pro 37.35, Agilent Technologies, Yarnton, UK, 2014 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- G. M. Sheldrick, SADABS-2008/1 - Bruker AXS Area Detector Scaling and Absorption Correction, Bruker AXS, Madison, Wisconsin, USA, 2008 Search PubMed.
- A. L. Spek, PLATON, A Multipurpose Crystallographic Tool 1.17, Utrecht University, Utrecht, The Netherlands, 2013 Search PubMed.
- SCM ADF, Theor. Chem, Vrije Univ. Amsterdam, Netherlands, http://www.scm.com Search PubMed.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- E. van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS PubMed.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef CAS.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783–9792 CrossRef CAS.
- A. Klamt, J. Phys. Chem., 1995, 99, 2224–2235 CrossRef CAS.
- A. Klamt and V. Jonas, J. Chem. Phys., 1996, 105, 9972–9981 CrossRef CAS.
- C. C. Pye and T. Ziegler, Theor. Chem. Acc., 1999, 101, 396–408 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 33, 8822–8824 CrossRef.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- J. R. Shakirova, E. V. Grachova, A. A. Melekhova, D. V. Krupenya, V. V. Gurzhiy, A. J. Karttunen, I. O. Koshevoy, A. S. Melnikov and S. P. Tunik, Eur. J. Inorg. Chem., 2012, 4048–4056 CrossRef CAS.
- T. M. Dau, J. R. Shakirova, A. Domenech, J. Jänis, M. Haukka, E. V. Grachova, T. A. Pakkanen, S. P. Tunik and I. O. Koshevoy, Eur. J. Inorg. Chem., 2013, 4976–4983 CAS.
- J. R. Shakirova, E. V. Grachova, A. S. Melnikov, V. V. Gurzhiy, S. P. Tunik, M. Haukka, T. A. Pakkanen and I. O. Koshevoy, Organometallics, 2013, 32, 4061–4069 CrossRef CAS.
- I. O. Koshevoy, Y. C. Chang, Y. A. Chen, A. J. Karttunen, E. V. Grachova, S. P. Tunik, J. Jänis, T. A. Pakkanen and P. T. Chou, Organometallics, 2014, 33, 2363–2371 CrossRef CAS.
- H.-Yi Chao, W. Lu, Y. Li, M. C. W. Chan, C.-M. Che, A. Kung-Kai Cheung and N. Zhu, J. Am. Chem. Soc., 2002, 124, 14696–14706 CrossRef CAS PubMed.
- J. Vicente, J. Gil-Rubio, N. Barquero, P. G. Jones and D. Bautista, Organometallics, 2008, 27, 646–659 CrossRef CAS.
- J. R. Shakirova, E. V. Grachova, V. V. Gurzhiy, I. O. Koshevoy, A. S. Melnikov, O. V. Sizova, S. P. Tunik and A. Laguna, Dalton Trans., 2012, 41, 2941–2949 RSC.
- J. R. Shakirova, E. V. Grachova, A. A. Melekhova, D. V. Krupenya, V. V. Gurzhiy, A. J. Karttunen, I. O. Koshevoy, A. S. Melnikov and S. P. Tunik, Eur. J. Inorg. Chem., 2012, 4048–4056 CrossRef CAS.
- J. R. Shakirova, E. V. Grachova, A. S. Melnikov, V. V. Gurzhiy, S. P. Tunik, M. Haukka, T. A. Pakkanen and I. O. Koshevoy, Organometallics, 2013, 32, 4061–4069 CrossRef CAS.
- I. O. Koshevoy, Y.-C. Chang, Y.-A. Chen, A. J. Karttunen, E. V. Grachova, S. P. Tunik, J. Jänis, T. A. Pakkanen and P.-T. Chou, Organometallics, 2014, 33, 2363–2371 CrossRef CAS.
- J. Camara, O. Crespo, M. C. Gimeno, I. O. Koshevoy, A. Laguna, I. Ospino, E. S. Smirnova, S. P. Tunik and T. A. Pakkanen, Dalton Trans., 2012, 41, 13891 RSC.
- M. Kadarkaraisamy, G. Caple, A. R. Gorden, M. A. Squire and A. G. Sykes, Inorg. Chem., 2008, 47, 11644–11655 CrossRef CAS PubMed.
- D. J. Hutchinson, M. P. James, L. R. Hanton and S. C. Moratti, Inorg. Chem., 2014, 53, 2122–2132 CrossRef CAS PubMed.
- R. Echeverria, J. M. Lopez-de-Luzuriaga, M. Monge and M. E. Olmos, Chem. Sci., 2015, 6, 2022–2026 RSC.
- R. Echeverria, J. M. Lopez-de-Luzuriaga, M. Monge, S. Moreno and M. E. Olmos, Inorg. Chem., 2016, 55, 10523–10534 CrossRef CAS PubMed.
- J. Wong, M. Todd and P. Rutledge, Molecules, 2017, 22, 200 CrossRef PubMed.
- C.-W. Hsu, C.-C. Lin, M.-W. Chung, Y. Chi, G.-H. Lee, P.-T. Chou, C.-H. Chang and P.-Y. Chen, J. Am. Chem. Soc., 2011, 133, 12085–12099 CrossRef CAS PubMed.
- W. Lu, W. Kwok, C. Ma, C. T. Chan and M. Zhu, J. Am. Chem. Soc., 2011, 133, 14120–14135 CrossRef CAS PubMed.
- Y. Chang, K. Tang, H. Pan, I. O. Koshevoy, A. J. Karttunen and P. Chou, J. Phys. Chem. C, 2013, 117, 20494–20499 CAS.
- E. Yu-Tzu Li, T.-Y. Jiang, Y. Chi and P.-T. Chou, Phys. Chem. Chem. Phys., 2014, 16, 26184–26192 RSC.
- I. Kondrasenko, K. Chung, Y.-T. Chen, J. Koivistoinen, E. V. Grachova, A. J. Karttunen, P.-T. Chou and I.O. Koshevoy, J. Phys. Chem. C, 2016, 120, 12196–12206 CAS.
- G. A. Crosby, R. G. Highland and K. A. Truesdell, Coord. Chem. Rev., 1985, 64, 41–52 CrossRef CAS.
- S. L. Zheng and X. M. Chen, Aust. J. Chem., 2004, 57, 703–712 CrossRef CAS.
- A. Barbieri, G. Accorsi and N. Armaroli, Chem. Commun., 2008, 2185–2193 RSC.
- Z. R. Ranjbar, A. Morsali and L.-G. Zhu, J. Coord. Chem., 2007, 60, 667–676 CrossRef CAS.
- M. Harvey, S. Baggio, R. Baggio, A. Mombrú and IUCr, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, 1457–1460 Search PubMed.
- Y. Hanifehpour, S. M. Asl, B. Mirtamizdoust and S. W. Joo, J. Inorg. Organomet. Polym. Mater., 2012, 22, 923–928 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1570478–1570481. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qi00514h |
| This journal is © the Partner Organisations 2018 |