
Conjugate substitution and addition of α-substituted acrylate: a highly efficient, facile, convenient, and versatile approach to fabricate degradable polymers by dynamic covalent chemistry†
 *a
*a
Abstract
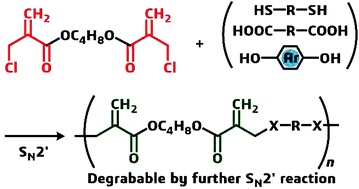
Polycondensation of bis[α-(halomethyl)acrylate] and dithiols, dicarboxylic acids, primary monoamines, and bisphenols via conjugate substitution yielded a series of poly(conjugated ester)s with acryloyl groups in the backbones. In the case of a dithiol monomer, a polar solvent promoted the conjugate addition (Michael addition) of a mercapto chain end into the acryloyl group in the resulting backbone to cause gelation. Therefore, kinetic control of the polymerization by the selection of nonpolar solvents and end capping with the mercapto group in the case of polar dithiols was necessary to prepare linear polymers. The monomer, bis[α-(halomethyl)acrylate], was also effective in achieving chain extension of common polyesters with phenolic hydroxyl end groups. These poly(conjugated ester)s underwent main chain scission when they were treated with benzyl mercaptan in the presence of a basic catalyst through a thiol exchange reaction via conjugate addition and substitution.
- This article is part of the themed collection: Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

