
Synthesis of a helical π-conjugated polymer with a dynamic hydrogen-bonded network in the helical cavity and its circularly polarized luminescence properties†

Abstract
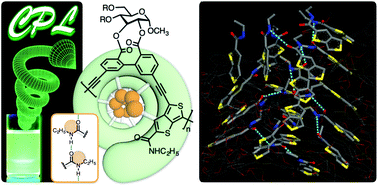
An optically active π-conjugated polymer (poly-1) containing glucose-linked biphenyl units in the main chain was synthesized via polymerization using an amide-appended 4,6-diiodothieno[3,4-b]thiophene as a cross-coupling partner. Based on the characteristic chiroptical properties of poly-1, combined with the result of all-atom molecular dynamics simulation, a cooperative intramolecular hydrogen-bonded network among amide pendants was considered to form in the helical cavity and to play a crucial role in the stabilization of the helically folded state. The helical poly-1 efficiently emitted circularly polarized light upon photoirradiation in solution and the film state.
- This article is part of the themed collection: Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

