
Polymer brush guided templating on well-defined rod-like cellulose nanocrystals†

Abstract
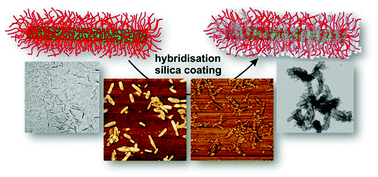
Cellulose is a natural biomaterial harvested from regrowing resources and it is one of the most attractive components for the construction of functional materials with low adverse ecological impact. Among various nanocelluloses, cellulose nanocrystals (CNC) are rod-like nanoparticles whose high crystallinity and stiffness make them viable candidates for templating materials. We report here on CNC-based polymer brushes used as templates for the synthesis of porous inorganic nanorods with tunable diameters and aspect ratios. The CNC were modified with initiation sites for surface-initiated polymerisation (SI-ATRP) to act as a backbone for the grafting of poly(2-(dimethyl amino)ethyl methacrylate) (PDMAEMA) brushes. Controlled polymerisation conditions allowed for adjusting the brush length and consequently the morphology of the hybrid nanomaterials. The PDMAEMA brush served as coordination and nucleation sites for the mineralisation of tetramethyl orthosilicate (TMOS) into SiO2@CNC-g-PDMAEMA hybrids. After calcination, microscopy and N2-sorption measurements revealed hollow silica nanorods with accessible micro- and meso-pores. We foresee that this strategy can be adapted to other nanocelluloses to create high-aspect ratio porous silica nanotubes, or to achieve uniform depositions in the mineralisation of other inorganic metals or metal oxide compounds.
- This article is part of the themed collection: Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

