DOI:
10.1039/C7PY01807J
(Paper)
Polym. Chem., 2018,
9, 48-59
Ethylene/propylene copolymerization catalyzed by half-titanocenes containing monodentate anionic nitrogen ligands: effect of ligands on catalytic behaviour and structure of copolymers†
Received
27th October 2017
, Accepted 25th November 2017
First published on 4th December 2017
Abstract
Various half-titanocenes containing monodentate anionic nitrogen ligands, CpTiCl2[3-C6H5(CH2N)(CH2O)C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N] (T1), CpTiCl2[(C6H5O)(NiPr2)CN] (T2), CpTiCl2[1,3-(4-MeC6H4)(CH2N)2CN] (T3), CpTiCl2[1,3-(2,4,6-Me3C6H2)(CH2N)2CN] (T4), CpTiCl2[1,3-(2,6-iPr2C6H3)(CH2N)2CN] (T5) and CpTiCl[1,3-(2,6-iPr2C6H3)(CH2N)2CN][CH3(CH2)3CH(C2H5)COO] (T6), were synthesized and characterized. The molecular structures of T1, T2 and T5 were determined by X-ray crystallography. All the above half-titanocenes were employed as precatalysts for ethylene/propylene copolymerization after activation by methylaluminoxane (MAO) or modified MAO (MMAO). The catalytic activity and propylene incorporation into copolymers increased in the order of T1 < T2 < T3 < T4 < T5, which is attributed to the electron donating nature of the ligands. The half-titanocene containing the 2-ethylhexanoate ligand (T6) has good solubility in hexanes. An ethylene–propylene copolymer with high molecular weight (Mw = 530 kg mol−1) could be obtained by the T6/MMAO catalytic system. The ethylene/propylene copolymerization catalyzed by the T6/TIBA/MMAO catalytic system was more controllable than that by the T5/MAO catalytic system. Importantly, both the T5/MAO and T6/TIBA/MMAO catalytic systems displayed a high catalytic activity of 2.4 × 106 and 2.2 × 106 g polymer per mol of Ti per h, respectively, even at a low Al/Ti molar ratio (Al/Ti = 300). All the values of rE × rP (rE: reactivity ratio for ethylene; rP: reactivity ratio for propylene) were around 0.9 in the copolymerization catalyzed by T2, T4, T5 and T6, indicating that nearly random ethylene–propylene copolymers could be prepared. Compared to the T5/MAO catalytic system, ethylene–propylene copolymers with higher molecular weight (Mw = 381 kg mol−1) and higher propylene incorporation (43.6 mol%) could be produced by using the T6/TIBA/MMAO (TIBA/Ti = 50) system with a similar catalytic activity.
N] (T1), CpTiCl2[(C6H5O)(NiPr2)CN] (T2), CpTiCl2[1,3-(4-MeC6H4)(CH2N)2CN] (T3), CpTiCl2[1,3-(2,4,6-Me3C6H2)(CH2N)2CN] (T4), CpTiCl2[1,3-(2,6-iPr2C6H3)(CH2N)2CN] (T5) and CpTiCl[1,3-(2,6-iPr2C6H3)(CH2N)2CN][CH3(CH2)3CH(C2H5)COO] (T6), were synthesized and characterized. The molecular structures of T1, T2 and T5 were determined by X-ray crystallography. All the above half-titanocenes were employed as precatalysts for ethylene/propylene copolymerization after activation by methylaluminoxane (MAO) or modified MAO (MMAO). The catalytic activity and propylene incorporation into copolymers increased in the order of T1 < T2 < T3 < T4 < T5, which is attributed to the electron donating nature of the ligands. The half-titanocene containing the 2-ethylhexanoate ligand (T6) has good solubility in hexanes. An ethylene–propylene copolymer with high molecular weight (Mw = 530 kg mol−1) could be obtained by the T6/MMAO catalytic system. The ethylene/propylene copolymerization catalyzed by the T6/TIBA/MMAO catalytic system was more controllable than that by the T5/MAO catalytic system. Importantly, both the T5/MAO and T6/TIBA/MMAO catalytic systems displayed a high catalytic activity of 2.4 × 106 and 2.2 × 106 g polymer per mol of Ti per h, respectively, even at a low Al/Ti molar ratio (Al/Ti = 300). All the values of rE × rP (rE: reactivity ratio for ethylene; rP: reactivity ratio for propylene) were around 0.9 in the copolymerization catalyzed by T2, T4, T5 and T6, indicating that nearly random ethylene–propylene copolymers could be prepared. Compared to the T5/MAO catalytic system, ethylene–propylene copolymers with higher molecular weight (Mw = 381 kg mol−1) and higher propylene incorporation (43.6 mol%) could be produced by using the T6/TIBA/MMAO (TIBA/Ti = 50) system with a similar catalytic activity.
Introduction
Ethylene–propylene rubber (EPR) and ethylene–propylene–diene rubber (EPDM) have an important market share of the industry due to their excellent resistance toward heat, oxidation, weather, and other chemicals.1–4 EPR and EPDM have been synthesized by ethylene/propylene copolymerization and ethylene/propylene/nonconjugated diene copolymerization, respectively, employing Ziegler–Natta2,4–6 and metallocene catalysts.2,4 The metallocene catalyst, as a single-site catalyst, becomes a promising choice because of its high activity, high thermal stability and excellent copolymerization ability.2,4 Nonbridged half-metallocenes containing monodentate anionic ligands have been widely investigated for ethylene/α-olefin (including propylene) copolymerization due to their better comonomer incorporation.7 In the reported nonbridged half-metallocenes, the monodentate anionic nitrogen ligands have drawn much attention.8–27 As shown in Scheme 1, the nonbridged half-metallocenes containing ketimide (a, b),8–12 amidinate (c),13–17 guanidinate (d),18,19 iminoimidazolidine (e, f, g)19–25 and phosphinimide (h)25–27 have been synthesized and used as precatalysts for ethylene homo- and co-polymerization with α-olefin. The ketimide half-metallocene (a) displayed high catalytic activity for ethylene polymerization and ethylene/1-hexene copolymerization.11,12 If the cyclopentadiene (Cp) ligand was replaced by the indenyl ligand, the activity of the ketimide half-metallocene (b) increased obviously for ethylene/1-hexene copolymerization.11 When one carbon atom connecting to the carbon atom in the CN group in the ketimide ligand was replaced by the nitrogen atom, the amidinate ligand was designed and used to prepare a half-metallocene precatalyst (c in Scheme 1).13–17 The half-sandwich amidinate titanium displayed high activity for ethylene/propylene/nonconjugated diene copolymerization.16 Using this type of precatalyst, an EPDM product marketed under the trade name Keltan ACE (“advanced catalysis elastomers”) could be produced in the copolymerization of ethylene, propylene and 5-ethylidene-2-norbornene (ENB).7,16 The precatalyst was further improved by introducing another nitrogen atom, resulting in half-titanocenes bearing guanidinate ligands (for example: d in Scheme 1).18 Half-titanocenes containing iminoimidazolidine were highly efficient precatalysts for ethylene (co)polymerization.19–25 Activated by B(C6F5)3, CpTi(CH2Ph)2[1,3-(2,6-Me2C6H3)2(CH2N)2CN] (e, X = CH2Ph) exhibited higher ethylene polymerization activity than its ketimide analogues (CpTi(CH2Ph)2(NCtBu2)).19 The catalytic behaviour was greatly affected by the substitution of the R group (f, R = tBu, C6H11, C6H5 and 2,6-Me2C6H3). The catalytic activity increased in the order of R: C6H11 < C6H5 < 2,6-Me2C6H3 < tBu in ethylene/1-hexene copolymerization. If the Cp ligand was replaced by the η5-pentamethylcyclopentadiene (Cp*) ligand (g),20–25 notable ethylene/1-hexene copolymerization activity was obtained using Cp*TiCl2[1,3-(2,6-Me2C6H3)2(CH2N)2CN] (g, R = Me) and Cp*TiCl2[1,3-(2,6-iPr2C6H3)2(CH2N)2CN] (g, R = iPr), respectively. However, a large amount of MAO was needed in these catalytic systems, i.e. the Al/Ti molar ratio ranged from 1 × 105 to 6 × 106.20 CpTi(CH3)2[1,3-(2,6-Me2C6H3)2(CH2N)2CN] (e, X = CH3) was an efficient precatalyst for the ethylene/propylene/ENB/VNB copolymerization.25 The half-titanocenes containing phosphinimide ligands (h) displayed similar catalytic behaviour toward ethylene/propylene/ENB/VNB copolymerization to the half-titanocenes containing iminoimidazolidine ligands.25–27 However, few studies have focused on the copolymerization of ethylene and propylene with the half-titanocenes containing iminoimidazolidine ligands.25 Especially, the effect of the ligand on the catalytic behaviour and microstructure of the ethylene–propylene copolymer has never been investigated.
 |
| | Scheme 1 The reported half-titanocenes containing monodentate anionic nitrogen ligands for ethylene (co)polymerization.8–27 | |
In this paper, the half-titanocenes containing 2-imino-3-phenyloxazolidine (T1), N,N-diisopropyl-O-(phenyl)isourea ligand (T2) or iminoimidazolidine and carboxylate ligands (T6) have been synthesized for the first time. The catalytic behaviour of these half-titanocenes together with the reported half-titanocenes containing iminoimidazolidine ligands (T3–T5) in ethylene/propylene copolymerization and the effect of the ligand on the microstructure of the resulting copolymer were investigated.
Results and discussion
Synthesis and characterization of half-titanocenes containing monodentate anionic nitrogen ligands
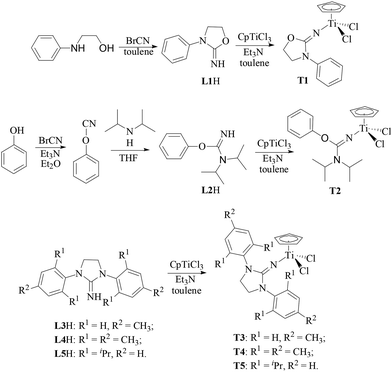
As shown in Scheme 2, 2-imino-3-phenyloxazolidine (L1H) could be obtained by the reaction of N-phenylamino ethanol with equivalent cyanogen bromide in toluene at 40 °C. The N,N-diisopropyl-O-(phenyl)isourea (L2H) was successfully synthesized for the first time by the reaction of phenylcyanate with equivalent diisopropylamine in THF. The imidazolin-2-iminato (L3H–L5H) ligands with various substitutes were prepared according to the reported method. A series of half-titanocenes containing L1–L5 could be synthesized with high yield by the reaction of equivalent CpTiCl3 and corresponding ligands in the presence of Et3N in toluene solution at 50 °C. The chemical structure of T1 and T2 was confirmed by 1H NMR spectroscopy, 13C NMR characterization and elemental analysis. The chemical structure of T3–T5 was characterized by 1H NMR and 13C NMR spectroscopy, which is the same as the reported results of half-titanocenes prepared by the reaction of CpTiCl3 and the corresponding imidazolin-2-iminato lithium salt.20,21
 |
| | Scheme 2 Synthesis of half-titanocenes containing monodentate anionic nitrogen ligands. | |
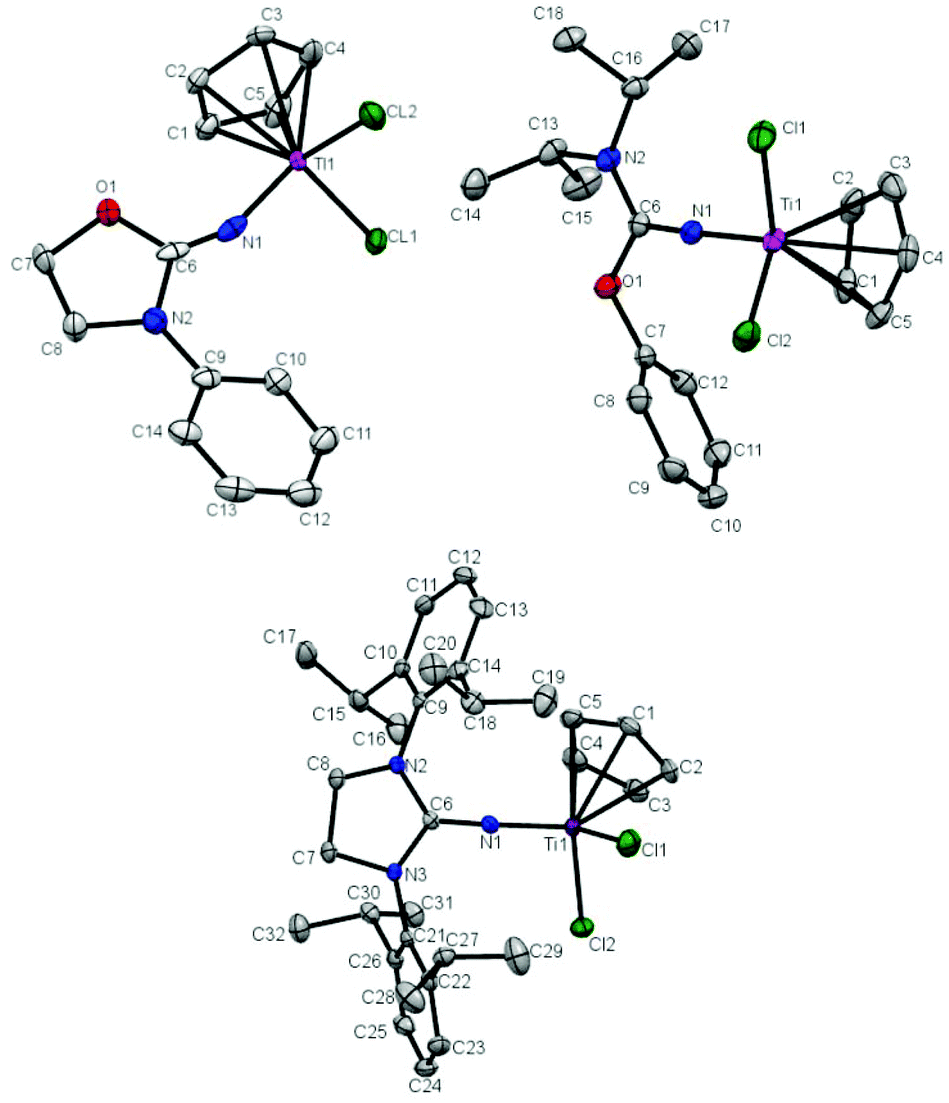
The molecular structure of T1, T2 and T5 was further determined by X-ray crystallography. Crystals of T1, T2 and T5 suitable for single-crystal X-ray analysis were grown by keeping their saturated toluene solution at −30 °C for 1 day. The molecular structures of T1, T2 and T5 are shown in Fig. 1 and the selected bond lengths and angles are summarized in Table 1. T1, T2 and T5 show a distorted tetrahedral geometry around titanium. The Ti–N bond distance in T1 (1.889 Å) is longer than that in T2 (1.7941 Å) and T5 (1.7849 Å), and the CN double bond distance in T1 (1.165 Å) is shorter than that in T2 (1.297 Å) and T5 (1.310 Å). The similar Ti–Cl bond distances about 2.30 Å in T1, T2 and T5 were measured. The distances between the center of Cp and Ti were determined to be 2.072 Å, 2.044 Å and 2.026 Å in T1, T2 and T5, respectively. The Ti–N–C bond angle in T1 (153.5°) was much narrower than that in T2 (171.14°) and T5 (174.44°), which indicates an inefficient ligand-to-metal π-donation in T1.20,21,23,24 The broad Cl–Ti–Cl angles in T1 (102.64°), T2 (100.41°) and T5 (101.57°) indicate a more open coordination environment for the monomer.
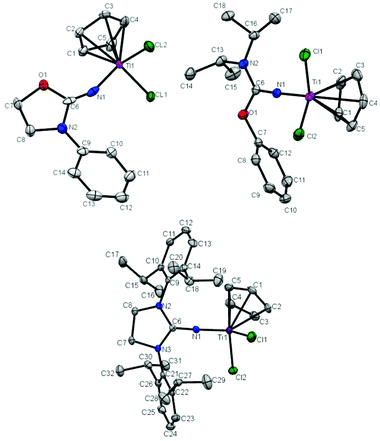 |
| | Fig. 1 Molecular structures of T1 (top left), T2 (top right) and T5 in T5·0.5C7H8 (bottom). Thermal ellipsoids are drawn at 30% probability. Hydrogen atoms are omitted for clarity. | |
Table 1 Selected bond distances (Å) and angles (°) for T1, T2 and T5
| |
T1
|
T2
|
T5
|
| Bond distances (Å) |
| Ti(1)–Cl(1) |
2.302 |
2.3027 |
2.2969 |
| Ti(1)–Cl(2) |
2.303 |
2.2980 |
2.3061 |
| Ti(1)–N(1) |
1.889 |
1.7941 |
1.7849 |
| N(1)–C(6) |
1.165 |
1.297 |
1.310 |
| Ti(1)–C(1) |
2.342 |
2.3536 |
2.3810 |
| Ti(1)–C(3) |
2.352 |
2.3734 |
2.430 |
| Ti(1)–Cp |
2.072 |
2.044 |
2.026 |
| |
| Bond angles (°) |
| Cl(1)–Ti(1)–Cl(2) |
102.64 |
100.41 |
101.57 |
| Ti(1)–N(1)–C(6) |
153.5 |
171.14 |
174.44 |
| Cl(1)–Ti(1)–N(1) |
100.77 |
101.76 |
103.79 |
| Cl(2)–Ti(1)–N(1) |
105.34 |
105.27 |
103.44 |
| Cp–Ti(1)–N(1) |
123.24 |
116.70 |
116.84 |
Effects of monodentate anionic nitrogen ligands on catalytic behaviour
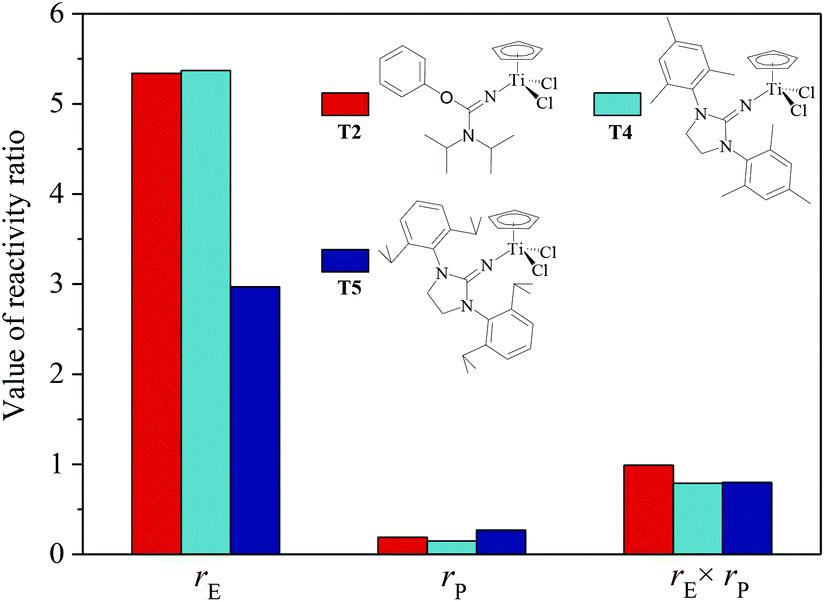
The ethylene/propylene copolymerizations using T1–T5/MAO catalytic systems were carried out in hexanes at 80 °C. The copolymerization results are displayed in Fig. 2 and 3. Activated by MAO (Al/Ti molar ratio = 1000), T1 containing the 2-imino-3-phenyloxazolidine (L1) ligand had much lower catalytic activity for ethylene/propylene copolymerization, which is attributed to the poor electron donating nature of the L1 ligand. Comparably, the T2/MAO and T3/MAO catalytic systems exhibited moderate catalytic activities of 8.0 × 104 and 1.6 × 105 g polymer per mol of Ti per h, respectively, for ethylene/propylene copolymerization due to the richer electron donating nature of L2 and L3. For T3, T4 and T5 containing imidazolin-2-iminato ligands with different substituents on the N-aryl-ring, the catalytic activity and propylene molar content in resulting ethylene–propylene copolymers increased in the order of T3 < T4 < T5. The similar trend of electronic effect on catalytic activity was also observed in the ethylene/1-hexene copolymerization catalyzed by half-titanocenes.24 The higher activity and propylene incorporation of the T5/MAO catalytic system could originate from the rather strong electron donating nature (more N → Ti donation) of ligand with isopropyl substitute on the N-aryl-ring. Compared to the catalytic activity of ethylene/1-hexene copolymerization using CpTiCl2[1,3-(C6H5)(CH2N)2CN] or CpTiCl2[1,3-(2,4,6-Me3C6H3)(CH2N)2CN] as precatalysts,24 the half-titanocenes of T4 and T5 exhibited slightly higher catalytic activity for ethylene/propylene copolymerization. Reactivity ratios of rE and rP were estimated from 13C NMR characterization according to the reported literature28 and the results are shown in Fig. 4. The high reactivity ratio of ethylene (rE = 5.34 and 5.37) and the low reactivity ratio of propylene (rP = 0.15 and 0.19) were obtained in the copolymerizations of ethylene with propylene with T2/MAO and T4/MAO catalytic systems. Compared to these two catalytic systems, the T5/MAO catalytic system exhibited a lower reactivity ratio of ethylene (rE = 2.97) and a higher reactivity ratio of propylene (rP = 0.27), respectively. The values of rE × rP were calculated to be 0.90 ± 0.1, indicating that nearly random ethylene–propylene copolymers could be prepared by the T2/MAO, T4/MAO and T5/MAO catalytic systems. The microstructures of the ethylene–propylene copolymers prepared by the T2/MAO, T4/MAO and T5/MAO catalytic systems were further investigated by 13C NMR characterization. Peaks were assigned in the spectra according to the reported literature.29–32 The triad fractions were calculated by the integral area of characteristic peaks according to the reported method.32 As shown in Fig. 5, the high fraction of the PPP triad and low fraction of the EEE triad could be observed from the copolymer prepared by the T5/MAO catalytic system, due to the decreased rE and increased rP in the copolymerization.
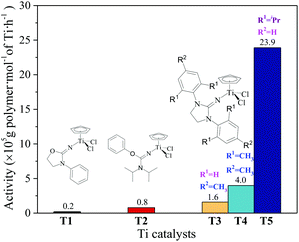 |
| | Fig. 2 The catalytic activity of the T1–T5/MAO catalytic systems in ethylene/propylene copolymerization. ([M] = 140 g L−1, P/E = 2 (molar ratio), [Ti]/[M] = 1.3 × 10−6, Al/Ti = 1000 (molar ratio), T = 80 °C, tp = 30 min.) The weights of the polymers: T1: 50 mg; T2: 210 mg; T3: 410 mg; T4: 990 mg; T5: 5980 mg. The viscosity-average molecular weight (Mη) of the polymers: T3: 4.3 × 105; T4: 4.6 × 105; T5: 3.4 × 105. The monomer conversion is given in Fig. S1, ESI.† | |
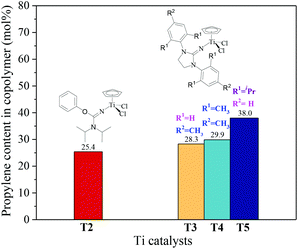 |
| | Fig. 3 The propylene content of ethylene–propylene copolymers prepared by the T1–T5/MAO catalytic systems. (Polymerization conditions are shown as in Fig. 2). | |
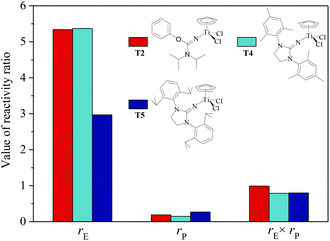 |
| | Fig. 4 The reactivity ratio of ethylene (rE) and propylene (rP) for the T2/MAO, T4/MAO and T5/MAO catalytic systems. | |
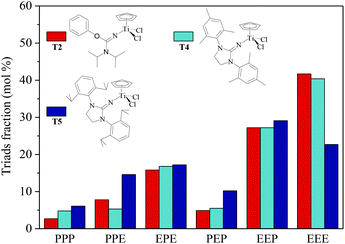 |
| | Fig. 5 Triad fraction of ethylene–propylene copolymers prepared by the T2/MAO, T4/MAO and T5/MAO catalytic systems. (Polymerization conditions are shown as in Fig. 2). | |
Fourier transformed infrared spectroscopy (FTIR) is also an effective method for microstructure characterization of the ethylene–propylene copolymer. The bands around 810 cm−l, 733 cm−l and 722 cm−l in the FTIR spectrum of the ethylene–propylene copolymer were ascribed to CH2, (CH2)3 and (CH2)n (n ≥ 5), respectively.33,34 The absorbance around 1157 cm−l was ascribed to the methyl group in the EPE triad33 and at 1460 cm−l could be ascribed to CH2 and CH3 bending bands. The absorbance at 810 cm−l, 733 cm−l, 722 cm−l and 1157 cm−l was divided by the absorbance at 1460 cm−1 in order to obtain normalized FTIR spectra. Then the peak intensity ratios of A810/A1460, A733/A1460, A722/A1460 and A1157/A1460 could be assigned to the PP, PEP, EEE + EEP and EPE sequence, respectively. The FTIR spectra of the resulting copolymers prepared by the T2/MAO and T5/MAO catalytic systems are shown in Fig. 6. A weaker band around 722 cm−1 and a stronger band around 1157 cm−1 in the FTIR spectrum of the copolymer prepared by the T5/MAO catalytic system indicate that more sequences with random distribution exist in the ethylene–propylene copolymer obtained by the T5/MAO catalytic system.
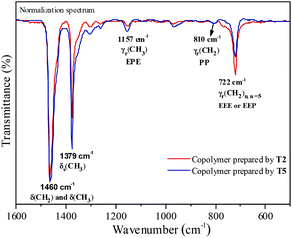 |
| | Fig. 6 Representative FTIR spectra of ethylene–propylene copolymers prepared by the T2/MAO and T5/MAO catalytic systems. (Polymerization conditions are shown as in Fig. 2). | |
Synthesis, characterization and catalytic behaviour of T6 containing the 2-ethylhexanoate ligand
T6 containing 2-ethylhexanoate was synthesized by the reaction of equivalent T5 with 2-ethylhexanoic acid in the presence of Et3N in toluene solution at 50 °C (as shown in Scheme 3). The chemical structure of T6 was characterized by 1H NMR and 13C NMR spectroscopy and elemental analysis. Different from T5 with extremely poor solubility in hexanes, T6 had good solubility in hexanes. MMAO in n-heptane was selected as a cocatalyst to avoid the contamination of toluene from MAO solution in the copolymerization system in hexanes.
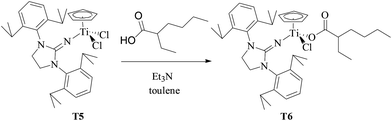 |
| | Scheme 3 Synthesis of T6 containing the 2-ethylhexanoate ligand. | |
The catalytic behaviour of T5 and T6 in the presence of MMAO in ethylene/propylene copolymerization was further investigated and the experimental results are given in Fig. 7. It can be observed from Fig. 7 that the ethylene–propylene copolymers exhibited unimodal molecular weight distribution and the polydispersities (Mw/Mn) were 2.2 and 2.3, respectively. This suggests that a uniform active species existed in polymerization with these catalytic systems. Moreover, as shown in Fig. 7, the weight-average molecular weight (Mw) of the resulting copolymer prepared by the T6/MMAO catalytic system was much higher than that prepared by the T5/MMAO catalytic system, suggesting that the introduction of the 2-ethylhexanoate substituent had an advantage of preparing the ethylene–propylene copolymer with high molecular weight. The average number of polymer chains (ncalcd) obtained by the T5/MMAO (7.0 × 10−5 mol) and T6/MMAO (9.2 × 10−6 mol) catalytic systems were calculated by mp/Mn, where mp is the weight of the copolymer obtained and Mn is number-average molecular weight of the copolymer (kg mol−1). The theoretical numbers of polymer chains (ntheo) in the copolymerization system catalysed by the catalytic systems were 5.0 × 10−6 mol. It can be observed that ncalcd was 14.1 times ntheo for the copolymerization system catalysed by the T5/MMAO catalytic system, which is attributed to serious chain-transfer to a cocatalyst during ethylene/propylene copolymerization. On the other hand, it can be also clearly seen that ncalcd was 1.8 times ntheo for the copolymerization system catalysed by the T6/MMAO catalytic system, which might be attributed to relatively low catalytic efficiency and/or less chain-transfer reaction to a cocatalyst during ethylene/propylene copolymerization. T6 might be activated for ethylene/propylene copolymerization by the elimination of one chlorine atom and one carboxylate group, which is similar to the mechanism in the reported titanium complexes containing a Ti–O bond activated by MAO for ethylene polymerization via a coordination–insertion pathway by the elimination of two isopropoxide groups.35
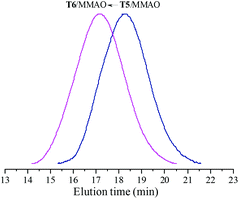 |
| | Fig. 7 The GPC traces of copolymers prepared by the T5/MMAO and T6/MMAO catalytic systems. (Polymerization conditions: [M] = 140 g L−1, P/E = 2 (molar ratio), [Ti]/[M] = 1.3 × 10−5, Al/Ti = 1000 (molar ratio), T = 50 °C, tp = 30 min.) (Polymerization results: T5/MMAO: activity = 2.4 × 106 g polymer per mol of Ti per h, P. content = 38.1 mol%, Mw = 185 kg mol−1, Mw/Mn = 2.2; T6/MMAO: activity = 7.6 × 105 g polymer per mol of Ti per h, P. content = 24.4 mol%, Mw = 530 kg mol−1, Mw/Mn = 2.3.). | |
The existence of a 2-ethylhexanoate ligand in T6 resulted in lower catalytic activity and propylene incorporation during ethylene/propylene copolymerization. In order to improve the catalytic activity of the T6/MMAO catalytic system, triisobutylaluminum (TIBA) was introduced to promote the alkylation reaction of T6. The mixture of T6 and TIBA with various TIBA/Ti molar ratios was added to the monomer solution containing MMAO to initiate the copolymerization. The catalytic activity and the composition of the ethylene–propylene copolymer prepared by the T6/TIBA/MMAO catalytic system at various TIBA/Ti molar ratios are shown in Fig. 8. The catalytic activity increased significantly from 7.6 × 105 to 2.3 × 106 g polymer per mol of Ti per h and the propylene contents of the resulting copolymer remarkably increased from 24.4 mol% to 45.3 mol% with an increase in the TIBA/Ti molar ratio from 0 to 100. The molecular weights of the resulting copolymers prepared by T6/TIBA/MMAO with various TIBA/Ti molar ratios were analyzed by GPC and the corresponding values of ncalcd were also calculated (as show in Fig. 9). The molecular weights of the resulting copolymers decreased and the corresponding values of ncalcd increased with TIBA/Ti molar ratio. The results turn out that the catalytic efficiency could be greatly improved and the chain-transfer reaction to a cocatalyst could be promoted in the presence of TIBA during ethylene/propylene copolymerization. Importantly, the ethylene–propylene copolymer with higher molecular weight (381 kg mol−1) and higher propylene content (43.6 mol%), having similar yield, could be synthesized by using the novel T6/TIBA/MMAO (TIBA/Ti = 50) catalytic system, compared to the T5/MAO catalytic system.
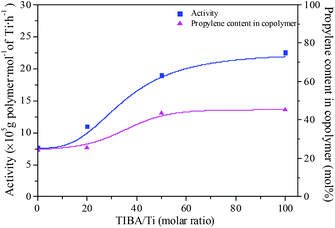 |
| | Fig. 8 The catalytic activity and the composition of the ethylene–propylene copolymer prepared by the T6/TIBA/MMAO catalytic system at various TIBA/Ti molar ratios. ([M] = 140 g L−1, P/E = 2 (molar ratio), [Ti]/[M] = 1.3 × 10−5, Al/Ti = 1000 (molar ratio), T = 50 °C, tp = 30 min. The monomer conversion is given in Fig. S2, ESI†.). | |
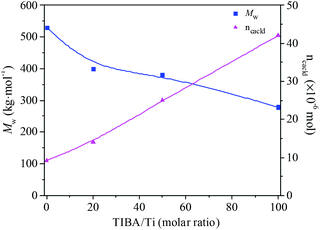 |
| | Fig. 9 The Mw of the ethylene–propylene copolymer and number of polymer chains (ncacld) of the T6/TIBA/MMAO catalytic system at various TIBA/Ti molar ratios. (Polymerization conditions are shown as in Fig. 8). | |
The FTIR spectra of the ethylene–propylene copolymer prepared by the T5/MAO and T6/TIBA/MMAO (TIBA/Ti = 50) catalytic systems are shown in Fig. 10. The A1157/A1460 (EPE) of the copolymer prepared by the T6/TIBA/MMAO (TIBA/Ti = 50) catalytic system was higher than that prepared by the T5/MAO catalytic system, which is attributed to higher propylene incorporation. The reactivity ratio rE and rP was also estimated based on 13C NMR spectroscopy (as shown in Fig. S3, ESI†). Compared to the T5/MAO catalytic system, the T6/TIBA/MMAO (TIBA/Ti = 50) catalytic system exhibited a similar reactivity ratio of ethylene (rE = 3.02) and slightly higher activity ratio of propylene (rP = 0.30), respectively. The value of rE × rP was determined to be 0.91. The results indicate that the ethylene/propylene copolymer with high propylene incorporation and random sequence distribution could be obtained by the T6/TIBA/MMAO (TIBA/Ti = 50) catalytic system.
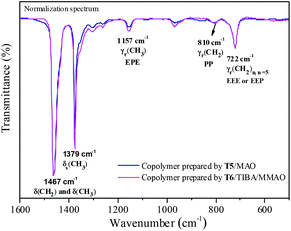 |
| | Fig. 10 Representative FTIR spectra of ethylene–propylene copolymers prepared by the T5/MAO and T6/TIBA/MMAO catalytic systems. (T5/MAO: [M] = 140 g L−1, P/E = 2 (molar ratio), [Ti]/[M] = 1.3 × 10−5, Al/Ti = 1000 (molar ratio), T = 50 °C, tp = 30 min. T6/MAO: TIBA/Ti = 50 (molar ratio); the other polymerization conditions are shown as in Fig. 8.). | |
Effect of copolymerization conditions on the catalytic behaviour of T5/MAO and T6/TIBA/MMAO catalytic systems
Al/Ti molar ratio.
MAO and MMAO are usually used as cocatalysts of metallocenes for olefin coordination polymerization. The primary function of MAO or MMAO is the activation of the metallocene catalyst. Therefore, a large excess amount of MAO or MMAO is needed in order to form the active centers and the Al/metal molar ratio has a significant influence on catalyst performance such as catalytic activity. The effect of Al/Ti molar ratio on catalytic activity and propylene content of the resulting copolymer are shown in Fig. 11 and 12. For the T5/MAO catalytic system, the maximum catalytic activity (2.4 × 106 g polymer per mol of Ti per h) was observed at a Al/Ti molar ratio of 300 or 500. The catalytic activity decreased slightly to 2.1 × 106 g polymer per mol of Ti per h with an increase in the Al/Ti molar ratio from 500 to 1500. At a low Al/Ti molar ratio, the amount of cocatalyst was insufficient to form active cationic species or to eliminate catalyst impurities present in the polymerization medium.7,36 An excess of MAO leads to a high concentration of trimethylaluminium associated with aluminoxane oligomers which could compete with the olefin for the vacant coordination site of the cationic metallocene.9,37,38 The composition of the resulting copolymers is not affected by the Al/Ti molar ratio. The ethylene–propylene copolymers with a propylene content of about 42.0 mol% were obtained at Al/Ti molar ratios between 300 and 1500. The sequences of the resulting copolymer were investigated by FTIR characterization (as shown in Fig. S5, ESI†). The results show that the amount of cocatalyst had little effect on the sequence distribution in the resulting ethylene–propylene copolymers.
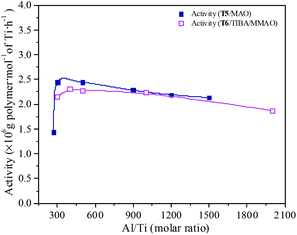 |
| | Fig. 11 The catalytic activity of the T5/MAO and T6/TIBA/MMAO catalytic systems at various Al/Ti molar ratios. (T5/MAO: [M] = 140 g L−1, P/E = 2 (molar ratio), [Ti]/[M] = 1.3 × 10−5, T = 80 °C, tp = 30 min; T6/TIBA/MMAO: [M] = 140 g L−1, P/E = 2 (molar ratio), [Ti]/[M] = 1.3 × 10−5, TIBA/Ti = 100, T = 50 °C, tp = 30 min. The monomer conversion is given in Fig. S4, ESI†.). | |
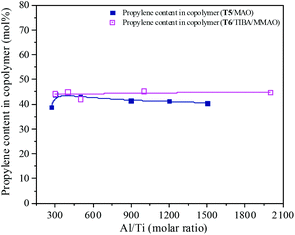 |
| | Fig. 12 The composition of the ethylene–propylene copolymer prepared by the T5/MAO and T6/TIBA/MMAO catalytic systems at various Al/Ti molar ratios. (Polymerization conditions are shown as in Fig. 11). | |
Similarly, the T6/TIBA/MMAO catalytic system also displayed high activity (2.2 × 106 g polymer per mol of Ti per h) at very low Al/Ti molar ratio (Al/Ti = 300), and reached a maximum value of 2.3 × 106 g polymer per mol of Ti per h when the Al/Ti molar ratio was 400 (as shown in Fig. 12). The Al/Ti molar ratio also had little effect on the composition and sequence of the resulting copolymers. The copolymers with propylene content about 45.0 mol% were obtained at Al/Ti molar ratios between 300 and 2000. The propylene contents of the resulting copolymers prepared by the T6/TIBA/MMAO catalytic system were higher than that prepared by the T5/MAO catalytic system.
Polymerization temperature.
Polymerization temperature plays an important role in copolymerization since it affects the solubility of monomer, solution viscosity, stability of the active center and reaction rate constant. Ethylene/propylene copolymerizations catalyzed by the T5/MAO and T6/TIBA/MMAO catalytic systems at various polymerization temperatures were investigated. In order to avoid strong exothermic effects, low catalyst concentrations ([Ti]/[M] = 4.7 × 10−6 for the T5/MAO catalytic system and [Ti]/[M] = 5.3 × 10−6 for the T6/TIBA/MMAO catalytic system) were used to keep the polymerization temperature from rising less than 15 °C. The copolymerization results are displayed in Fig. 13 and 14, respectively. It can be clearly observed from Fig. 13 that the catalytic activity of the T5/MAO catalytic system increased firstly when the initial polymerization temperature was increased from 2.5 to 60 °C and reached a maximum value of 5.9 × 106 g polymer per mol of Ti per h at 60 °C. Then the catalytic activity decreased to 5.3 × 106 g polymer per mol of Ti per h with an increase in the initial polymerization temperature from 60 to 80 °C. This trend could be ascribed to the competition between the decrease of monomer concentration, deactivation of catalyst and the increase of propagation rate with an increase in polymerization temperature. A different behaviour of the T6/TIBA/MMAO catalytic system was observed at various polymerization temperatures. As shown in Fig. 13, with increasing initial polymerization temperature from 15 to 90 °C, the catalytic activity firstly increased from 5.0 × 106 to 6.4 × 106 g polymer per mol of Ti per h and then decreased gradually to 3.6 × 106 g polymer per mol of Ti per h. The maximum catalytic activity was observed at an initial polymerization temperature of 30 °C. The T6/TIBA/MMAO catalytic system displayed higher catalytic activity than the T5/MAO catalytic system at a low initial polymerization temperature. The composition of the resulting copolymer was also dependent obviously on the polymerization temperature. In general, the insertion of propylene always requires higher activation energy than the insertion of ethylene. Therefore, the insertion of propylene should become easier with increasing polymerization temperature.39,40 However, as shown in Fig. 14, the ethylene–propylene copolymers with lower propylene content were obtained at higher temperature for both the T5/MAO and T6/TIBA/MMAO catalytic systems, which could be corresponding to the faster rotation of bulky isopropyl groups at higher temperature. The rotation of the bulky isopropyl substitute occupies more space around the metal center and hence hinders propylene insertion for chain propagation.39 The influence of initial polymerization temperature on the triad fractions of the resulting copolymer was investigated based on the 13C NMR characterization (as shown in Fig. S7, ESI†). It reveals that the resulting copolymers with lower EEE triad fraction and higher EPE + PEP triad fractions could be prepared at lower initial polymerization temperature. A similar trend was also observed by FTIR characterization and the results are shown in the ESI† Fig. S8. The sequence in the resulting copolymer prepared by the T6/TIBA/MMAO catalytic system was investigated based on FTIR characterization. The detailed results are shown in the ESI† Fig. S9. The intensity ratios of A722/A1460 (EEE + EEP) increased and A1157/A1460 (EPE) decreased with an increase in the initial polymerization temperature. This trend is similar to the results for the T5/MAO catalytic system. The variation of sequence at different initial polymerization temperatures indicates that the ethylene–propylene copolymers containing more sequences with random distribution could be synthesized with the T5/MAO and T6/TIBA/MMAO catalytic systems at a low initial polymerization temperature.
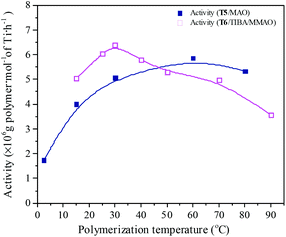 |
| | Fig. 13 The catalytic activity of the T5/MAO and T6/TIBA/MMAO catalytic systems at various initial polymerization temperatures. (T5/MAO: [M] = 80 g L−1, P/E = 2 (molar ratio), [Ti]/[M] = 4.7 × 10−6, Al/Ti = 2000 (molar ratio), tp = 30 min; T6/TIBA/MMAO: [M] = 140 g L−1, P/E = 2 (molar ratio), [Ti]/[M] = 5.3 × 10−6, TIBA/Ti = 100, Al/Ti = 1000 (molar ratio), tp = 30 min. The monomer conversion is given in Fig. S6, ESI†.). | |
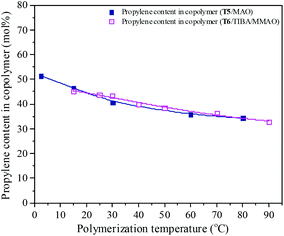 |
| | Fig. 14 The composition of the ethylene–propylene copolymer prepared by the T5/MAO and T6/TIBA/MMAO catalytic systems at various initial polymerization temperatures. (Polymerization conditions are shown as in Fig. 13.). | |
Polymerization time.
Ethylene/propylene copolymerization was carried out with various polymerization times from 1 min to 30 min by the T5/MAO and T6/TIBA/MMAO catalytic systems under their optimum polymerization conditions. As shown in the ESI† Fig. S10, the temperature in polymerization solution with the T5/MAO catalytic system rose rapidly from 60 to 65 °C within the initial 1 min, and reached a maximum of 67 °C in 2 min. Then the temperature of the reaction system decreased with further prolonging the polymerization time. The temperature in the polymerization solution with the T6/TIBA/MMAO catalytic system increased gradually from 30 °C to 45 °C within 17 min. As shown in Fig. 15 and 16, the conversion–time curves reflect the rapid increase of conversion of monomer for the T5/MAO catalytic system and gradual increase of conversion of monomer for the T6/TIBA/MMAO catalytic system. It seems that the copolymerization of ethylene with propylene by the T6/TIBA/MMAO catalytic system was more controllable than that by the T5/MAO catalytic system.
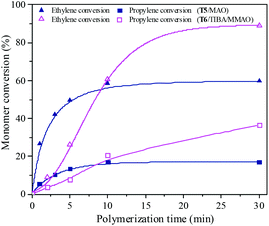 |
| | Fig. 15 The ethylene and propylene conversion at various polymerization times by using the T5/MAO and T6/TIBA/MMAO catalytic systems. (T5/MAO: [M] = 80 g L−1, P/E = 2 (molar ratio), [Ti]/[M] = 4.7 × 10−6, Al/Ti = 2000 (molar ratio), T = 60 °C; T6/TIBA/MMAO: [M] = 140 g L−1, P/E = 2 (molar ratio), [Ti]/[M] = 5.3 × 10−6, TIBA/Ti = 100, Al/Ti = 1000 (molar ratio), T = 30 °C). | |
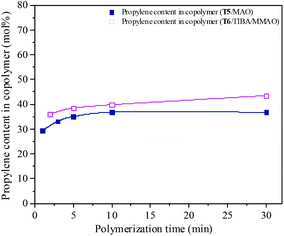 |
| | Fig. 16 The composition of the ethylene–propylene copolymer prepared by the T5/MAO and T6/TIBA/MMAO catalytic systems at various polymerization times. (Polymerization conditions are shown as in Fig. 15.). | |
The propylene contents of the resulting copolymers prepared by the T6/TIBA/MMAO catalytic system increased from 36.1 to 43.5 mol% by prolonging the polymerization time from 2 min to 30 min. Whereas, copolymers with lower propylene contents from 29.4 to 36.8 mol% were obtained by the T5/MAO catalytic system by prolonging polymerization time from 1 min to 30 min. It can be inferred that the ethylene–propylene copolymers with higher propylene contents could be achieved by using the T6/TIBA/MMAO catalytic system.
Conclusions
The desired half-titanocenes containing various monodentate anionic nitrogen ligands (T1–T6) could be successfully synthesized and employed as precatalysts for ethylene/propylene copolymerization. The catalytic behaviour of these half-titanocenes and the microstructure of the resulting ethylene–propylene copolymers could be mediated by the chemical structure of the ligands. The T2/MAO and T3/MAO catalytic systems exhibit higher catalytic activity due to their richer electron donating nature than the T1/MAO catalytic system for ethylene/propylene copolymerization. For the half-titanocenes T3, T4 and T5 containing imidazolin-2-iminato ligands with different substituents on the N-aryl-ring, the catalytic activity and propylene molar content in the resulting ethylene–propylene copolymers increased in the order of T3 < T4 < T5 due to an increase in the electron donating nature (more N → Ti donation) of the ligand. The half-titanocenes of T1–T5 with two Cl− ligands could be dissolved in toluene but could not be dissolved in hexanes. T6 with good solubility in hexanes could be obtained by the replacement of one Cl− ligand in T5 by the 2-ethylhexanoate ligand. An ethylene–propylene copolymer with a high molecular weight (Mw = 530 kg mol−1) could be obtained by the T6/MMAO catalytic system. The catalytic activity of the T6/MMAO system could be further improved by the introduction of TIBA. Both the T5/MAO and T6/TIBA/MMAO catalytic systems exhibited a high catalytic activity of 2.4 × 106 and 2.2 × 106 g polymer per mol of Ti per h, respectively, even at a low Al/Ti molar ratio (Al/Ti = 300). The copolymerization of ethylene with propylene by the T6/TIBA/MMAO catalytic system was more controllable than that by the T5/MAO catalytic system. Interestingly, with catalytic activity similar to the T5/MAO catalytic system, the ethylene–propylene copolymer prepared by using the T6/TIBA/MMAO catalytic system exhibits a higher molecular weight (Mw = 381 kg mol−1), a propylene content of 43.6 mol% and more sequences with random distribution. The results should be valuable to the design of half-metallocenes for ethylene/propylene copolymerization and the preparation of the ethylene–propylene copolymer with high molecular weight and more sequences with random distribution.
Experimental section
General procedure
All experiments were carried out under a nitrogen atmosphere drybox or using standard Schlenk techniques. Toluene (Beijing Chemical Works) was distilled under nitrogen atmosphere and refluxed over sodium benzophenone for dehydration, and then stored in the drybox in the presence of molecular sieves (4A). Hexanes (Beijing Chemical Works) were stored in the drybox with molecular sieves (4A) after refluxing over calcium hydride. Ethylene and propylene (Beijing ZG Special Gases Science and Technology Co. Ltd) were used as received without further purification. MAO was a commercial solution in toluene (Albemarle, 10% w/w). MMAO was a commercial solution in n-heptane (Akzo Nobel, 10% w/w). All 1H and 13C NMR spectra were recorded on Bruker AV 400 MHz NMR spectrometers operating at 25 °C (400 MHz for 1H and 100 MHz for 13C). Chemical shifts are given in ppm and are referenced to SiMe4 (δ 0.00, 1H, 13C). Elemental analyses (C, H, N) were recorded by vario EL cube Elementar Analysensysteme GmbH. Fourier transform infrared spectroscopy (FTIR) measurements were performed on a Nicolet 6700 infrared spectrometer equipped with a DTGS detector at ambient temperature by using samples prepared in potassium bromide (KBr). For each spectrum, a 24-scan interferogram was collected in the single beam mode with a 4 cm−1 resolution and a 1.928 cm−1 interval from 2000 cm−1 to 400 cm−1 regions. The ethylene and propylene contents of the ethylene–propylene copolymer were calculated according to ASTM D3900. The monomer conversion (Conv.%) was calculated by WM × mp/mM × 100%, where WM is the weight fraction of the monomer in the copolymer (wt%), mp is the weight of the copolymer obtained and mM is the weight of monomer added. Molecular weights (Mw and Mn) and molecular weight distributions (Mw/Mn) of the ethylene–propylene copolymer were determined using a PL-GPC220 at 150 °C with 1,2,4-trichlorobenzene as the solvent. Intrinsic viscosities of all samples were measured in 1,2,4-trichlorobenzene at 135 °C and the solution concentrations were 0.57 g dL−1. The viscosity-average molecular weights of the copolymer were obtained according to the reported method.41
Synthesis of 2-imino-3-phenyloxazolidine (L1H) and its half-titanocene T1
2-Imino-3-phenyloxazolidine was synthesized according to the reported synthetic method.42 The toulene solution (20 mL) of N-phenylamino ethanol (2.66 g, 19 mmol) was introduced into a two necked flask equipped with a reflux condenser and dropping funnel. Cyanogen bromide (2.00 g, 19 mmol) in toluene (20 mL) was added dropwise within 0.5 h at 0 °C. The solution was stirred for a further hour at 40 °C. The separated precipitate was filtered off, washed with diethyl ether and dried. The obtained white solid was dissolved in water. The solution was filtered and the filtrate was rendered strongly alkaline by introducing 10 N aqueous sodium hydroxide solution. The separated white precipitate was filtered off and solved in CHCl3. The solution was dried by MgSO4. The pure product 2-imino-3-phenyloxazolidine as a white solid (L1H, 1.68 g, 10.4 mmol, yield = 54%) was obtained after removing the solvent. 1H NMR (400 MHz, CDCl3), δ 4.01 (t, 2H, NCH2CH2O), 4.39 (t, 2H, NCH2CH2O), 5.49 (s, H, CNH), 7.09 (t, H, p-C6H5), 7.39 (t, 2H, m-C6H5), 7.63 (d, 2H, o-C6H5); 13C NMR (100 MHz, CDCl3), δ 47.06, 62.62, 118.78, 122.92, 128.73, 139.61, 157.69. Into a toluene solution (10 mL) containing CpTiCl3 (0.22 g, 1.0 mmol) was added a toluene solution (10 mL) of 2-imino-3-phenyloxazolidine (0.16 g, 1.0 mmol) and Et3N (0.15 mL, 1.0 mmol) at 50 °C. The reaction mixture was stirred for 5 h at 50 °C, and extracted by hot toluene. The extracts were concentrated in vacuum, and placed in a freezer. T1 was obtained as a yellow solid (0.29 g, 0.83 mmol, yield = 83.1%). 1H NMR (400 MHz, CDCl3), δ 4.24 (t, 2H, NCH2CH2O), 4.60 (t, 2H, NCH2CH2O), 6.59 (s, 5H, C5H5), 7.18 (t, H, p-C6H5), 7.42 (t, 2H, m-C6H5), 7.66 (d, 2H, o-C6H5). 13C NMR (100 MHz, CDCl3), δ 47.44, 64.02, 117.07, 120.24, 125.57, 129.31, 136.82, 150.26. Anal. calcd for C14H14Cl2N2OTi: C 48.73, H 4.09, N 8.12%. Found: C 49.09, H 4.29, N 8.22%.
Synthesis of N,N-diisopropyl-O-(phenyl)isourea and its half-titanocene T2
N,N-Diisopropyl-O-(phenyl)isourea was synthesized according to the reported synthetic method.43,44 To a diethyl ether solution (40 mL) of phenol (2.60 g, 27.5 mmol) and Et3N (3.10 g, 30.3 mmol) in a one necked flask equipped with a dropping funnel, cyanogen bromide (3.20 g, 30.0 mmol) in 10 mL of diethyl etherwas added dropwise within 0.5 h at 0 °C. The obtained solution was stirred for a further hour at room temperature. The separated precipitate was filtered off, and the pure product phenylcyanate as a white solid (2.62 g, 22.0 mmol, yield = 80%) was obtained after removing the solvent. Into a THF solution (15 mL) of phenylcyanate (1.00 g, 7.5 mmol) was added diisopropylamine (1.0 mL, 7.5 mmol), and the mixture was stirred for 10 h at room temperature; the pure product N,N-diisopropyl-O-(phenyl)isourea as a white solid (L2H, 1.15 g, 5.2 mmol, yield = 69%) was obtained after removing the solvent. 1H NMR (400 MHz, CDCl3), δ 1.30 (d, 12H, NiPr), 4.05 (m, 2H, (CH3)2CH), 7.10 (d, 2H, o-C6H5), 7.21 (t, H, p-C6H5), 7.39 (t, 2H, m-C6H5); 13C NMR(100 MHz, CDCl3), δ 21.05, 46.17, 121.76, 124.96, 129.50, 151.11, 159.60. T2 was synthesized in the same manner as that for T1 by the reaction of equivalent CpTiCl3 (0.50 g, 2.3 mmol) and N,N-diisopropyl-O-(phenyl)isourea (0.50 g, 2.3 mmol) in the presence of Et3N (0.30 mL, 2.0 mmol) at 50 °C. The resultant T2 was obtained as a yellow solid (0.71 g, 1.8 mmol, yield = 77.5%). 1H NMR (400 MHz, CDCl3), δ 1.25 (d, 6H, NiPr), 1.45 (d, 6H, NiPr), 3.64 (m, H, (CH3)2CH), 4.89 (m, H, (CH3)2CH), 6.06 (s, 5H, C5H5), 7.20 (d, 2H, o-C6H5), 7.32 (t, H, p-C6H5), 7.44 (t, 2H, m-C6H5); 13C NMR (100 MHz, CDCl3), δ 20.10, 21.63, 46.69, 50.61, 116.21, 122.72, 126.10, 129.58, 149.81, 151.14. Anal. calcd for C18H24Cl2N2OTi: C 53.62, H 6.00, N 6.95%. Found: C 54.67, H 6.47, N 6.82%.
Synthesis of half-titanocenes containing imidazolin-2-iminato ligands
A series of imidazolin-2-iminato (L3H to L5H) ligands were synthesized according to the reported procedure,19,24,45 and T3 to T5 were synthesized in the same manner as that for T1 which was different from the reported procedure.24T3 was synthesized by the reaction of equivalent CpTiCl3 (0.42 g, 1.9 mmol) and L3H (0.50 g, 1.9 mmol) in the presence of Et3N (0.29 mL, 1.9 mmol) at 50 °C. The resultant T3 was obtained as a yellow solid (0.34 g, 0.76 mmol, yield = 40.2%). 1H NMR (400 MHz, CDCl3), δ 2.35 (s, 6H, Ph–CH3), 4.08 (s, 4H, NCH2CH2N), 6.01 (s, 5H, C5H5), 7.21 (d, 4H, m-C6H4), 7.43 (s, 4H, o-C6H4); 13C NMR (100 MHz, CDCl3), δ 21.20, 49.32, 119.23, 125.08, 131.20, 132.60, 139.44, 154.82. Anal. calcd for C22H23Cl2N3Ti: C 58.95, H 5.17, N 9.38%. Found: C 59.46, H 5.23, N 8.98%; T4 was synthesized by the reaction of equivalent CpTiCl3 (0.36 g, 1.6 mmol) and L4H (0.51 g, 1.5 mmol) in the presence of Et3N (0.24 mL, 1.6 mmol) at 50 °C. The resultant T4 was obtained as a yellow solid (0.64 g, 1.3 mmol, yield = 79.3%). 1H NMR (400 MHz, CDCl3), δ 2.35 (s, 18H, Ph–CH3), 3.93 (s, 4H, NCH2CH2N), 5.92 (s, 5H, C5H5), 6.98 (s, 4H, m-C6H2); 13C NMR (100 MHz, CDCl3), δ 17.95, 21.08, 46.15, 115.01, 129.41, 132.40, 136.82, 138.62, 150.66. Anal. calcd for C26H31Cl2N3Ti·0.5C7H8: C 64.38, H 6.41, N 7.63%. Found: C 64.38, H 7.05, N 7.89%; T5 was synthesized by the reaction of equivalent CpTiCl3 (0.62 g, 2.8 mmol) and L5H (1.13 g, 2.8 mmol) in the presence of Et3N (0.42 mL, 2.8 mmol) at 50 °C. The resultant T5 was obtained as a yellow solid (1.22 g, 2.1 mmol, yield = 74.1%). 1H NMR (400 MHz, CDCl3), δ 1.33 (d, 12H, CH(CH3)2), 1.48 (d, 12H, CH(CH3)2), 3.22 (m, 4H, CH(CH3)2), 4.02 (s, 4H, NCH2CH2N), 5.84 (s, 5H, C5H5), 7.28 (d, 4H, m-C6H3), 7.42 (s, 2H, p-C6H3); 13C NMR (100 MHz, CDCl3), δ 23.72, 25.49, 48.65, 115.35, 124.37, 129.66, 132.68, 147.62, 150.48.
Synthesis of T6 half-titanocene containing 2-ethylhexanoate ligands
T6 was synthesized in the same manner as T1 by the reaction of equivalent T5 (0.40 g, 0.7 mmol) and 2-ethylhexanoic acid (0.10 g, 0.7 mmol) at 50 °C in the presence of Et3N (0.10 mL, 0.7 mmol). The resultant T6 was obtained as a brown oil (0.40 g, 0.6 mmol, yield = 83.1%). 1H NMR (400 MHz, CDCl3), δ 0.80–0.95 (m, 6H, CH3(CH2)3CH(CH2CH3)COO), 1.20–1.50 (m, 18H, CH3(CH2)3CH(CH2CH3)COO, CH(CH3)2), 1.61 (m, 2H, CH3(CH2)3CH(CH2CH3)COO), 2.07 (m, 1H, CH3(CH2)3CH(CH2CH3)COO), 3.11 (m, 4H, CH(CH3)2), 3.85 (s, 4H, NCH2CH2N), 6.52 (s, 5H, C5H5), 7.21 (d, 4H, m-C6H3), 7.25 (t, 2H, p-C6H3); 13C NMR (100 MHz, CDCl3), δ 11.96, 14.00, 22.76, 24.23, 24.41, 25.26, 28.84, 29.56, 30.60, 48.59, 48.72, 121.96, 124.41, 129.11, 133.34, 148.67, 159.66, 193.97. Anal. calcd for C40H58Cl2N3O2Ti·1/6C7H8: C 69.58, H 8.71, N 5.80%. Found: C 69.33, H 9.28, N 5.43%.
Crystallographic analysis
The crystallographic analyses were made on an Oxford Diffraction Gemini E diffractometer with Mo-Kα radiation (λ = 0.7107 Å) in the ω scan mode. All structures were solved by direct methods and refined by full-matrix least-squares refinement on F2 (SHELXL-97).46 The non-hydrogen atoms were refined anisotropically. The hydrogen atoms were added geometrically and refined with a mixed model. The selected crystal collection parameters are listed in Table 2 and the detailed results were described in the reports in the ESI.† CCDC reference numbers for T1, T2 and T5 are CCDC 1565929–1565931,† respectively.
Table 2 Crystal data and collection parameters of T1, T2 and T5·0.5C7H8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
| |
T1
|
T2
|
T5·0.5C7H8 |
|
Details in the crystallographic data for T1, T2 and T5: CCDC 1565929–1565931, respectively.
|
| Formula |
C14H14Cl2N2OTi |
C18H24Cl2N2OTi |
C32H43Cl2N3Ti·0.5C7H8 |
| Formula weight |
345.07 |
403.19 |
634.56 |
| Crystal color/habit |
Yellow/block |
Yellow/block |
Yellow/block |
| Crystal size (mm) |
0.14 × 0.23 × 0.55 |
0.35 × 0.34 × 0.30 |
0.34 × 0.4 × 0.45 |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
| Space group |
P21/c |
P21/c |
P21/n |
|
a (Å) |
15.3461 |
14.5895 |
18.8611 |
|
b (Å) |
9.6856 |
10.0182 |
19.4952 |
|
c (Å) |
20.5438 |
13.9424 |
19.5123 |
|
β (°) |
105.618 |
101.750 |
105.765 |
|
V (Å3) |
2940.8 |
1995.13 |
6904.8 |
|
Z value |
8 |
4 |
8 |
|
D
calcd(g cm−3) |
1.559 |
1.342 |
1.221 |
|
F(000) |
1408 |
840 |
2696 |
| Temp. (K) |
106.1 |
107.1 |
106.1 |
|
μ (mm−1) |
0.941 |
0.704 |
0.430 |
| Reflections collected |
15847 |
9083 |
15854 |
| Independent reflections |
5755 |
3913 |
13543 |
| Data/restraints/parameters |
5755/12/361 |
3913/0/221 |
13543/0/765 |
|
R
1(I > 2.00σ(I)) |
0.0870 |
0.0318 |
0.0369 |
| wR2(I > 2.00σ(I)) |
0.1932 |
0.0732 |
0.0832 |
| Goodness of fit |
1.204 |
1.048 |
1.021 |
| Largest diff. peak/hole (e Å−3) |
1.794/−0.660 |
0.281/−0.267 |
0.451/−0.366 |
| Completeness |
0.998 |
0.998 |
0.998 |
Procedure for ethylene/propylene copolymerization
Typical ethylene/propylene copolymerization (Fig. 2) was carried out as follows. Anhydrous hexanes (100 mL) were added into the autoclave (250 mL), followed by propylene (10.5 g) and ethylene (3.5 g) at room temperature. MAO in toluene (10% w/w, 3.0 mL) was added into the autoclave at 80 °C, and then a toluene solution (2.0 mL) containing the prescribed amount of titanium complexes (5 μmol) was added to start the copolymerization reaction. The copolymerization reactions were carried out for 30 min, and ethylene–propylene copolymers were obtained after the addition of HCl/ethanol (5% w/w) into the polymerization solution and filtration, and then dried in vacuum at 40 °C until a constant weight.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support from the National Natural Science Foundation of China (21634002; 51521062) is gratefully appreciated.
Notes and references
- S. Cesca, J. Polym. Sci., Macromol. Rev., 1975, 10, 1 CrossRef.
-
J. W. M. Noordermeer, Encyclopedia of Polymer Science and Technology, John Wiley & Sons, Inc, 2002, vol. 6, p. 178 Search PubMed.
- M. C. Haag, J. H. Z. Dos Santos, F. C. Stedile and J. Dupont, J. Appl. Polym. Sci., 1999, 74, 1997 CrossRef CAS.
- P. S. Ravishankar, Rubber Chem. Technol., 2012, 85, 327 CrossRef CAS.
- S. Gambarotta, Coord. Chem. Rev., 2003, 237, 229 CrossRef CAS.
- S. Zhang, W. C. Zhang, D. D. Shang, Z. Q. Zhang and Y. X. Wu, Dalton Trans., 2015, 44, 15264 RSC.
- M. C. Baier, M. A. Zuideveld and S. Mecking, Angew. Chem., Int. Ed., 2014, 53, 9722 CrossRef CAS PubMed.
-
M. John, X. L. Gao, S. R. E. von Haken, B. S. John and J. Dusan, WO, 9914250A1, 1999 Search PubMed.
- M. J. Ferreira and A. M. Martins, Coord. Chem. Rev., 2006, 250, 118 CrossRef CAS.
- A. R. Dias, M. T. Duarte, A. C. Fernandes, S. Fernandes, M. M. Marques, A. M. Martins, J. F. da Silva and S. S. Rodrigues, J. Org. Chem., 2004, 689, 203 CrossRef CAS.
- K. Nomura, K. Fujita and M. Fujiki, J. Mol. Catal. A: Chem., 2004, 220, 133 CrossRef CAS.
- K. Nomura and J. Y. Liu, Dalton Trans., 2011, 40, 7666 RSC.
- E. G. Ijpeij, B. Coussens, M. A. Zuideveld, G. H. J. van Doremaele, P. Mountford, M. Lutz and A. L. Spek, Chem. Commun., 2010, 46, 3339 RSC.
-
E. G. Ijpeij, P. J. H. Windmuller, A. H. Johannes, B. F. van Der, G. H. J. van Doremaele and M. A. Zuideveld, WO, 2005090418A1, 2005 Search PubMed.
-
E. G. Ijpeij, M. A. Zuideveld, H. J. Arts, B. F. van Der and G. H. J. van Doremaele, WO, 2007031295A1, 2007 Search PubMed.
- G. H. J. van Doremaele, M. van Duin, M. Valla and A. Berthoud, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2877 CrossRef CAS.
- R. A. Collins, A. F. Russell, R. T. W. Scott, R. Bernardo, G. H. J. van Doremaele, A. Berthoud and P. Mountford, Organometallics, 2017, 36, 2167 CrossRef CAS.
-
G. H. J. van Doremaele, M. A. Zuideveld, A. Leblanc and N. V. F. Quirogal, EP, 2319874A1, 2011 Search PubMed.
- W. P. Kretschmer, C. Dijkhuis, A. Meetsma, B. Hessen and J. H. Teuben, Chem. Commun., 2002, 6, 608 RSC.
- K. Nomura, S. Patamma, H. Matsuda, S. Katao, K. Tsutsumi and H. Fukuda, RSC Adv., 2015, 5, 64503 RSC.
- K. Nomura, H. Fukuda, H. Matsuda, S. Katao and S. Patamma, J. Org. Chem., 2015, 798, 375 CrossRef CAS.
- X. Wu and M. Tamm, Coord. Chem. Rev., 2014, 260, 116 CrossRef CAS.
- K. Nomura, H. Fukuda, W. Apisuk, A. G. Trambitas, B. Kitiyanan and M. Tamm, J. Mol. Catal. A: Chem., 2012, 363, 501 CrossRef.
- K. Nomura, H. Fukuda, S. Katao, Mi. Fujiki, H. J. Kim, D. H. Kim and S. Zhang, Dalton Trans., 2011, 40, 7842 RSC.
-
P. J. H. Windmuller and G. H. J. van Doremaele, WO2005005496A2, 2005 Search PubMed.
-
S. R. E. von Haken, D. W. Stephan, S. J. Brown, D. Jeremin and Q. Y. Wang, WO, 0005236, 2000 Search PubMed.
- D. W. Stephan, J. C. Stewart, F. Guérin, S. Courtenay, J. Kickham, E. Hollink, C. Beddie, A. Hoskin, T. Graham, P. R. Wei, R. E. v. H. Spence, W. Xu, L. Koch, X. L. Gao and D. G. Harrison, Organometallics, 2003, 22, 1937 CrossRef CAS.
- T. Uozumi and K. Soga, Makromol. Chem., 1992, 193, 823 CrossRef CAS.
- C. J. Carman, R. A. Harrington and C. E. Wilkes, Macromolecules, 1976, 10, 536 CrossRef.
- J. C. Randall, Macromolecules, 1978, 11, 33 CrossRef CAS.
- H. N. Cheng, Macromolecules, 1984, 17, 1950 CrossRef CAS.
- Y. Feng and J. N. Hay, Polymer, 1988, 39, 6589 CrossRef.
- H. V. Drushel and F. A. Iddings, Anal. Chem., 1963, 35, 28 CrossRef CAS.
- T. A. Veerkamp and A. Veermans, Makromol. Chem., 1961, 50, 147 CrossRef CAS.
- M. K. Panda, S. Kaur, A. R. Reddy, M. M. Shaikh, R. J. Butcher, V. Gupta and P. Ghosh, Dalton Trans., 2010, 39, 11060 RSC.
- W. Kaminsky and H. Sinn, Adv. Polym. Sci., 2013, 258, 1 CrossRef CAS.
- S. Ahmadjo, H. Arabi, M. Nekoomanesh, G. H. Zohuri, M. M. Mortazavi and G. Nader, Macromol. React. Eng., 2010, 4, 707 CrossRef CAS.
- M. Mortazavi, H. Arabi, S. Ahmadjo, M. Nekoomanesh and G. H. Zohuri, J. Appl. Polym. Sci., 2011, 122, 1838 CrossRef CAS.
- L. Lu, H. Niu, J. Y. Dong, X. T. Zhao and X. T. Hu, J. Appl. Polym. Sci., 2010, 118, 3218 CrossRef CAS.
- J. C. W. Chien and B. Xu, Makromol. Chem., Rapid Commun., 1993, 14, 109 CrossRef CAS.
- J. T. Gotro and W. W. Graessley, Macromolecules, 1984, 17, 2767 CrossRef CAS.
-
J. T. Thurston, US, 2479525A, 1949 Search PubMed.
- D. Martin and M. Bauer, Org. Synth., 1983, 61, 35 CrossRef CAS.
- K. Tsuchiya and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 699 CrossRef CAS.
-
L. Toldy, Z. Zubovics, M. Kürti and I. Schäfer, DE, 2916140A1, 1979 Search PubMed.
-
G. M. Sheldrick, SHELX-97, University of Göttingen, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Part of the copolymerization results and structure reports for half-titanocenes T1, T2 and T5. CCDC 1565929–1565931. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7py01807j |
|
| This journal is © The Royal Society of Chemistry 2018 |

 *
*