
Swelling properties of thermoresponsive/hydrophilic co-networks with functional crosslinked domain structures†
 *a
*a
Abstract
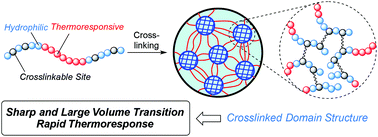
For the fine control of the thermoresponsive properties of polymer hydrogels, we focused on the monomer and crosslinker sequence in a network. In this paper, we designed novel thermoresponsive/hydrophilic polymer co-networks with functional crosslinked domain (CD) structures such as a thermoresponsive network with hydrophilic CDs (DCDN gel) and a hydrophilic network with thermoresponsive CDs (NCDD gel). The gel synthesis was based on post-polymerization crosslinking of triblock prepolymers with reactive sites in the outer blocks. The obtained gels showed larger, sharper, and more rapid volume transitions in response to temperature change as compared to the corresponding gel with a random monomer/crosslinker sequence as well as a randomly crosslinked polymer co-network with the same composition. In addition, significant differences were observed in the transition temperature and shrinking kinetics between DCDN and NCDD gels with the same composition and a similar network structure, and the swelling behavior was also controlled by the composition of the triblock prepolymers. These results indicated that the domain structure in the network could effectively function and the monomer/crosslinker sequence exerts a strong effect on the swelling behavior of the gels.
- This article is part of the themed collection: Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

