
Secondary structures of synthetic polypeptide polymers

Abstract
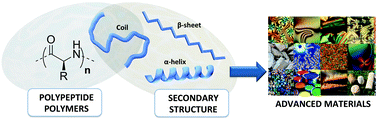
Synthetic peptide-based polymers represent a unique class of macromolecules able to mimic the properties of natural proteins in materials sciences since (1) they present the same macromolecular backbone as do proteins, (2) they can be obtained in large scale and in only one step by using the ROP (Ring-Opening Polymerization) methodology, and (3) they can fold into different secondary structures in the same way as do proteins. The control of this structuring ability paves the way to a wide range of applications in materials science, for which uses of natural proteins remain limited. In this review article, the fundamental principles of polypeptide polymer structuring are summarized. It is also highlighted here, how tuning the polypeptide secondary structure could be a key step to modulate various properties in advanced materials (size, rigidity, self-assembly, etc.).
- This article is part of the themed collection: Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

