
The influence of the substituents of oxiranes on copolymerization with vinyl ethers via concurrent cationic vinyl-addition and ring-opening mechanisms†
 *a
Sadahito
Aoshima
*a
*a
Sadahito
Aoshima
*a
Abstract
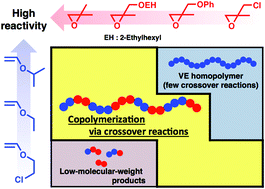
2,2-Disubstituted oxiranes with electron-withdrawing substituents were copolymerized with vinyl ethers (VEs) via concurrent cationic vinyl-addition and ring-opening mechanisms, leading to a design strategy for copolymerization via crossover reactions with high efficiency. β-Methylglycidyl ether (βMGE) and β-methylepichlorohydrin, which contain an alkoxy group and a chlorine atom, respectively, as electron-withdrawing groups, exhibited lower reactivity in copolymerization with VEs than isobutylene oxide, a dialkyl-substituted counterpart. Thus, VEs with appropriate reactivities depending on the reactivity of the oxirane were required. For example, 2-ethylhexyl βMGE (EHMGE) and phenyl βMGE ether were not efficiently consumed in the reaction with isopropyl VE but underwent copolymerization via crossover reactions with less reactive ethyl VE (EVE) or 2-chloroethyl VE. Specifically, the copolymerization of EVE and EHMGE yielded a copolymer with an Mn value of 7.5 × 103via 8.3 times crossover reactions from VE to βMGE per chain. The effects of oxirane substituents on the copolymerization behavior are discussed in terms of the reactivities of monomers and propagating species. In addition, a βMGE with an oxyethylenic chain was demonstrated to be effective for the generation of thermoresponsive copolymers.
- This article is part of the themed collection: Industry R&D collection
 Please wait while we load your content...
Please wait while we load your content...

