
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pure hydrophilic block copolymer vesicles with redox- and pH-cleavable crosslinks†
Jochen
Willersinn
and
Bernhard V. K. J.
Schmidt
 *
*
Max-Planck Institute of Colloids and Interfaces; Department of Colloid Chemistry, Am Mühlenberg 1, 14476 Potsdam, Germany. E-mail: Bernhard.schmidt@mpikg.mpg.de
First published on 22nd August 2017
Abstract
The self-assembly of a novel double hydrophilic block copolymer consisting of biocompatible blocks, namely pullulan-b-poly(N-vinylpyrrolidone), is presented. Completely hydrophilic spherical structures with an average apparent radius of 800 nm at increased concentrations in water are observed via dynamic light scattering as well as cryo scanning electron microscopy and confocal laser scanning microscopy techniques. Moreover, the pullulan block is converted to present aldehyde groups acting as anchor point for crosslinker attachments. It is demonstrated, that the oxidized self-assembled particles could be crosslinked via the bifunctional crosslinker cystamine forming dynamic covalent imine linkages with aldehyde groups. The afforded vesicles with an average diameter of 700 nm are stable upon high dilution and could be observed via cryo scanning electron microscopy and transmission electron microscopy. Furthermore, it is possible to cleave the crosslinking bonds with the treatment of acid or the application of a reducing agent. The responsivity is a key feature taking future applications in the biomedical sector into account.
Introduction
Block copolymer self-assembly has significant impact on polymer science as well as chemical science in general.1–3 Plenty of utilizations of block copolymer self-assemblies are found in various chemical disciplines, e.g. block copolymer templating in mesoporous silica synthesis,4,5 energy related materials6 or nano structure engineering.7,8 One of the most frequently utilized types of block copolymers for self-assembly are amphiphilic block copolymers that can be used for the formation of micelles,9–11 cylindrical micelles12,13 or vesicles14,15 in aqueous environment. Concomitantly colloidal structures from amphiphilic block copolymers have found broad interest with respect to applications, e.g. as drug delivery vehicles,14,15 hybrid materials,16 photoluminescent materials17 or membranes.18In contrast to the self-assembly of amphiphilic block copolymers in aqueous solution, pure hydrophilic block copolymers, e.g. double hydrophilic block copolymers (DHBCs), can form self-assembled structures in aqueous solution as well. On one hand stimuli-responsive blocks were employed to form such structures. Usually a stimulus like temperature or pH change is utilized to render the solubility of one block in the DHBC to hydrophobic.19 Certainly, a triggered self-assembly has several advantages but in this case the self-assembly is due to the hydrophobic effect and the block copolymer not a pure DHBC at the time of self-assembly. On the other hand self-assembly of pure DHBC in water without external triggers is possible as shown by several researchers, e.g. giant vesicles of pullulan-b-poly(ethylene oxide) (Pull-b-PEO),20 vesicles from Pull-b-poly(N-ethylacrylamide),21 particles from poly(2-ethyl-2-oxazoline)-b-poly(N-vinylpyrrolidone) (PEtOx-b-PVP),22 micelles from poly(2-hydroxyethyl methacrylate)-b-poly(2-O-(N-acetyl-β-D-glucosamine)ethyl methacrylate)23 or lyotropic mesophases from PEO-b-poly(2-methyl-2-oxazoline).24 Certainly, depending on the choice of the blocks DHBCs might feature significant biocompatibility.25
The synthesis of DHBCs can be performed via reversible deactivation radical polymerization strategies, e.g. the reversible addition-chain transfer fragmentation (RAFT)/macromolecular architecture design via the interchange of xanthates (MADIX) process,26 as well as modular conjugation reactions such as the copper catalyzed azide alkyne cycloaddition reaction.27 While the block copolymer formation is straightforward, the self-assembly process of DHBCs is not completely understood. Although there are theoretical investigations,28 the microstructure of the formed self-assemblies is still unknown. In general relatively high polymer concentrations are required for the formation of self-assembled structures, which renders them on the other hand unstable in dilute environment. In order to preserve the completely hydrophilic self-assemblies crosslinking in aqueous solution is a valid option as it has been utilized frequently in nano particle29,30 and hydrogel formation.31,32 In the case of DHBC-based systems crosslinking has been performed with self-assemblies from PEO-b-poly(N-vinylpyrrolidone) (PEO-b-PVP)33 and PEO-b-PEtOx.34
A bio-derived building block that has proven to be useful in DHBC self-assembly so far is Pull,20,21 which is a linear non-ionic poly(saccharide) consisting of α-1,6-linked maltotriose units.35 It entails significant biodegradability,35 biocompatibility36 and can be end functionalized easily.20,37 A combination with the likewise biocompatible PVP38 seems to be an efficient combination with respect to future application in biomedical science. In such a way a novel DHBC is generated that can be investigated regarding self-assembly processes in water (Scheme 1). According to research by Whitesides and coworkers, PVP and dextran homopolymers form multiphase systems in water.39 Therefore, a dextran-b-PVP block copolymer might form self-assemblies in aqueous solution, which is an indication that Pull-b-PVP forms self-assemblies as Pull is structurally related to dextran.
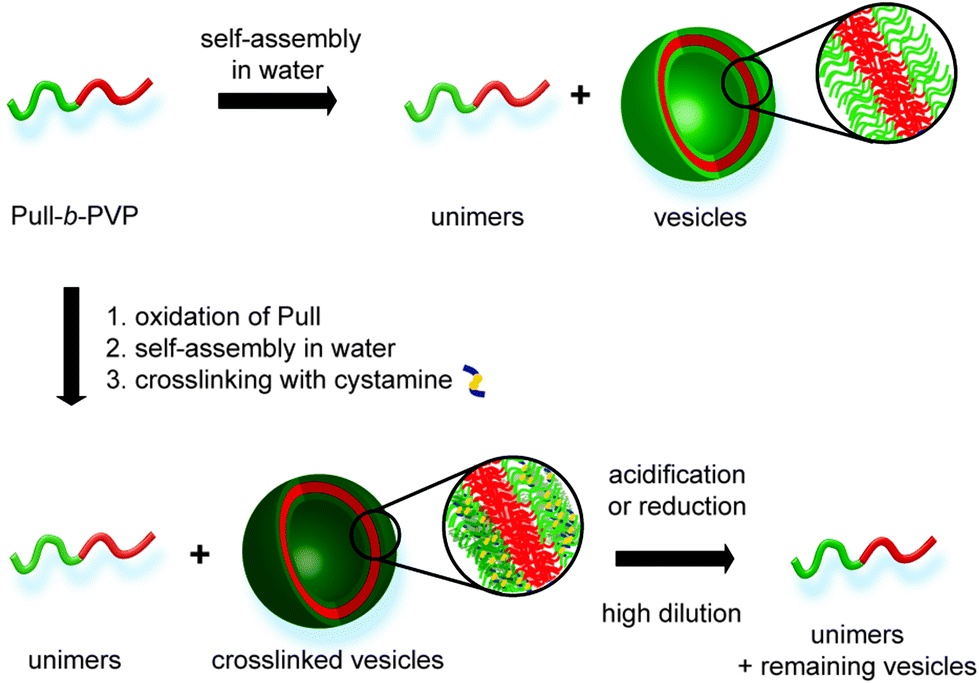 | ||
| Scheme 1 Overview for self-assembly of Pull-b-PVP, crosslinking via cystamine and pH or redox induced cleavage. | ||
As stated above, crosslinking of the formed self-assembled structure is beneficial for stabilization and a significant requirement for utilization in applicable concentration ranges. In that regard oxidation of the Pull backbone for the formation of aldehyde moieties allows crosslinking via the formation of pH responsive dynamic covalent imine bonds40,41 together with a diamine molecule, e.g. cystamine. Therefore, pH cleavable crosslinking is reached. Moreover, utilization of the functional diamine cystamine entails the system with another feature of triggered cleavage. As cystamine contains a disulfide bond, cleavage can be induced in reductive environment.42,43 In such a way DHBC-based crosslinked self-assemblies are obtained that can be cleaved via pH or redox triggers and intentionally disassembled at high dilution (Scheme 1).
Here a novel DHBC, namely Pull-b-PVP, undergoing self-assembly in aqueous solution is presented. The block copolymer is synthesized via a Pull macro chain transfer agent suitable for chain extension with VP via RAFT polymerization. Subsequently, the formation of self-assembled structures was probed via dynamic light scattering (DLS), cryo scanning electron microscopy (SEM), confocal laser scanning microscopy (CLSM) and differential interference contrast microscopy (DIC) showing particles in the range of 150 nm to 1 μm. Moreover, the Pull block could be oxidized and crosslinked via addition of cystamine. Therefore, stable structures could be generated and analyzed via DLS, cryo SEM and transmission electron microscopy (TEM). As shown via cryo SEM hollow spheres are obtained indicating vesicular structures in solution. Additionally, the formed crosslinks could be cleaved via pH due to the dynamic covalent nature of the imine bond or redox triggers due to the incorporated disulfide moiety leading to degraded vesicles at high dilution.
Experimental
Chemicals
N-Vinylpyrrolidone (VP, 99%, Sigma Aldrich) was dried over anhydrous magnesium sulfate and purified by distillation under reduced pressure. Acetone (analytical grade, J.T. Baker) and dichloromethane (DCM, analytical grade, Acros Organics) were stored over molecular sieves (3 Å) prior to use. Millipore water was obtained from an Integra UV plus pure water system by SG Water (Germany). Ammonium chloride (99%, Roth KG), 2-bromopropionyl bromide (97%, Sigma Aldrich), t-butyl peroxide (70% solution in water, Acros Organics), cystamine dihydrochloride (96%, Sigma Aldrich), diethylene glycol (99%, Fischer Chemical), diethyl ether (ACS reagent, Sigma Aldrich), N,N-dimethylformamide (DMF, analytical grade, Sigma Aldrich), dimethylsulfoxide (DMSO, analytical grade, VWR Chemicals), ethyl acetate (EtOAc, analytical grade, Chem Solute), hexamethylene diamine (96%, Sigma Aldrich), hexane (analytical grade, Fluka), hydrochloric acid (HCl, fuming, Roth KG), N-hydroxy succinimide (NHS, 98%, Sigma Aldrich), magnesium sulfate (dried, Fisher Scientific), methanol (MeOH, analytical grade, Fisher Scientific), potassium-O-ethyl xanthate (98% Alfa Aesar), pyridine (99% extra dry, Acros Organics), Rhodamine B (Sigma Aldrich), sodium bicarbonate (>99%, Fluka), sodium cyanoborohydride (NaCNBH3, 95%, Sigma Aldrich), sodium (meta)periodate (NaIO4, pure, VWR Chemicals), sodium sulfite (97%, Acros Organics), triethylamine (Sigma Aldrich) and tris(2-carboxyethyl)phosphine hydrochloride (TCEP, >98%, Roth KG) were used as received. Spectra/Por dialysis tubes with MWCOs of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 1000000 were purchased from Spectrum Labs. 2,5-Dioxopyrrolidin-1-yl-2-bromopropanoate was synthesized according to the literature (see ESI† for details).44 Pullulan was depolymerized and conjugated with an amine according to the literature (see ESI† for details).37,45
000 and 1000000 were purchased from Spectrum Labs. 2,5-Dioxopyrrolidin-1-yl-2-bromopropanoate was synthesized according to the literature (see ESI† for details).44 Pullulan was depolymerized and conjugated with an amine according to the literature (see ESI† for details).37,45
Analytical techniques
1H- and 13C-NMR spectra were recorded at ambient temperature at 400 MHz for 1H and 100 MHz for 13C with a Bruker Ascend400. Size exclusion chromatography (SEC) was conducted in N-methyl-2-pyrrolidone (NMP, Fluka, GC grade) with 0.05 mol L−1 LiBr and BSME as internal standard at 70 °C using a column system by PSS GRAM 100/1000 column (8 × 300 mm, 7 μm particle size) with a PSS GRAM precolumn (8 × 50 mm) and a Shodex RI-71 detector and a calibration with PEO standards from PSS. Pullulan samples were analyzed in acetate buffer containing 20% MeOH at 25 °C using a PSS NOVEMA Max analytical system XL (pre column size 50 mm × 8 mm – 10 μm, main column size 300 mm × 8 mm – 10 μm) using a pullulan calibration with standards from PSS. Dynamic light scattering (DLS) was performed using an ALV-7004 Multiple Tau Digital Correlator in combination with a CGS-3 Compact Goniometer and a HeNe laser (Polytec, 34 mW, λ = 633 nm in a θ = 90° setup to the photo-detector (ALV/SO-SIPD)). Sample temperatures were adjusted to 25 °C with a toluene bath surrounding the cuvette. Apparent radii (Rapp) were determined by DLS and the data were fitted using the REPES algorithm, which is similar to the CONTIN algorithm and suitable for multi modal distributions. Cryogenic scanning electron microscopy (cryo SEM) was performed with a Jeol JSM 7500 F and the cryo-chamber from Gatan (Alto 2500). Confocal laser scanning microscopy (CLSM) imaging was conducted with a Leica TCS SP5 (Wetzlar, Germany) confocal microscope, using a 63× (1.2 NA) water immersion objective. The dye stained samples were excited with a diode pumped solid-state laser at 561 nm and the emission bands were collected at 640 nm. Transmission electron microscopy (TEM) was performed with a Zeiss EM 912 Ω microscope operated at an acceleration voltage of 120 kV. The samples were cast on a carbon coated copper TEM grid and freeze dried for analysis.:2, Rf = 0.27) on silica gel to afford 2,5-dioxopyrrolidin-1-yl 2-((ethoxycarbonothioyl)thio)propanoate (NHS-xanthate) (1.1 g, 3.87 mmol, 42% yield). 1H NMR (400 MHz, CDCl3, δ:) 4.67 (dq, 2J1,2 = 2.3 Hz, 3J2,3 = 7.1 Hz, 2H, 2-H), 4.61 (q, 3J = 7.4 Hz, 1H, 4-H), 2.83 (s, 4H, 8-H, 9-H), 1.70 (d, 3J = 7.4 Hz, 3H, 5-H), 1.42 (t, 3J = 7.1 Hz, 3H, 1-H). 13C-NMR (100 MHz, 300 K, CDCl3, δ:) 13.5 (1-C), 16.5 (5-C), 25.6 (8-C, 9-C), 44.2 (4-C), 70.9 (2-C), 167.6 (7-C, 10-C), 168.7 (6-C), 209.9 (3-C).
000 MWCO Spectra Por) and lyophilized to afford Pull-b-PVP (1.47 g, 0.025 mmol) as white powder. Mn,app,SEC = 58800 g mol−1 (PEO equivalents in NMP), Đ = 1.77.
100 g mol−1, PEO equivalents in NMP, Đ = 2.1; 10% oxidation: 0.433 g, 0.011 mmol, Mn,app,SEC = 39200 g mol−1, PEO equivalents in NMP, Đ = 1.8) as a white powder.
1.9 μL (3.8 μL for the 10% oxidized block copolymer, respectively) of the stock solution was added to the Pull124-b-PVP263 solution and the reaction mixture was shaken for 2 days. The reaction mixture was diluted to 0.1 wt% (100 μL of crosslinked solution was added to 5 mL Millipore water) and dialyzed against Millipore water with a 1000000 MWCO dialysis tube for 3 days. The resulting solution was analyzed via DLS at 25 °C.
Results and discussion
Synthesis of ω-functionalized pullulan-xanthate
In order to synthesize a Pull macroinitiator for the block copolymer synthesis of Pull-b-PVP, natural Pull precursor was depolymerized according to the literature (see ESI for details, Fig. S1 and Table S1†). In a subsequent step the depolymerized Pull was end functionalized with hexamethylene diamine to introduce an amine functionalization (see ESI for details, Fig. S2 and S3†).After the successful transformation of the ω-aldehyde to a primary amine,37 the attachment of a RAFT/MADIX chain transfer agent was performed in the next step (Scheme S1†). Therefore, a N-succinimidyl xanthate, namely 2,5-dioxopyrrolidin-1-yl 2-((ethoxycarbonothioyl)thio)propanoate was synthesized via literature known procedures (Fig. S4 and S5†).44,46 The active ester route was chosen to avoid side reactions, e.g. aminolysis of the RAFT agent as well as multiple attachments. The synthesis of the pullulan chain transfer agent was carried out in DMSO at ambient temperature to ensure complete dissolution of pullulan amine and the xanthate transfer agent. The xanthate functionalized pullulan was dialyzed and lyophilized after the reaction to remove DMSO and unreacted xanthate transfer agent from the solution (Scheme S2†). An apparent average molecular weight of 26500 g mol−1 with Đ of 1.7 was obtained (Table S1 and Fig. S6†). 1H-NMR displays the successful attachment of the xanthate group to pullulan amine. The proton signals corresponding to the xanthate and the hexamethylene group could be assigned via the magnification of the area with a low chemical shift (Fig. S7†). Subsequently, block copolymer formation via chain extension with VP was performed.
Block copolymer formation of Pull-b-PVP
Block copolymer formation via RAFT polymerization of VP in aqueous solution according to our previous method33 led to unsatisfying results. Therefore, the system was diluted with another polar but aprotic solvent, namely DMSO (Fig. 1a). DMSO is a good solvent for monomer and macro RAFT/MADIX chain transfer agent. The polymerization was performed at 25 °C with the redox couple sodium sulfite/t-butyl hydroperoxide as initiator. A Pull124-b-PVP263 block copolymer with an apparent number weighted molecular mass of 58800 g mol−1 and Đ of 1.8 was obtained.
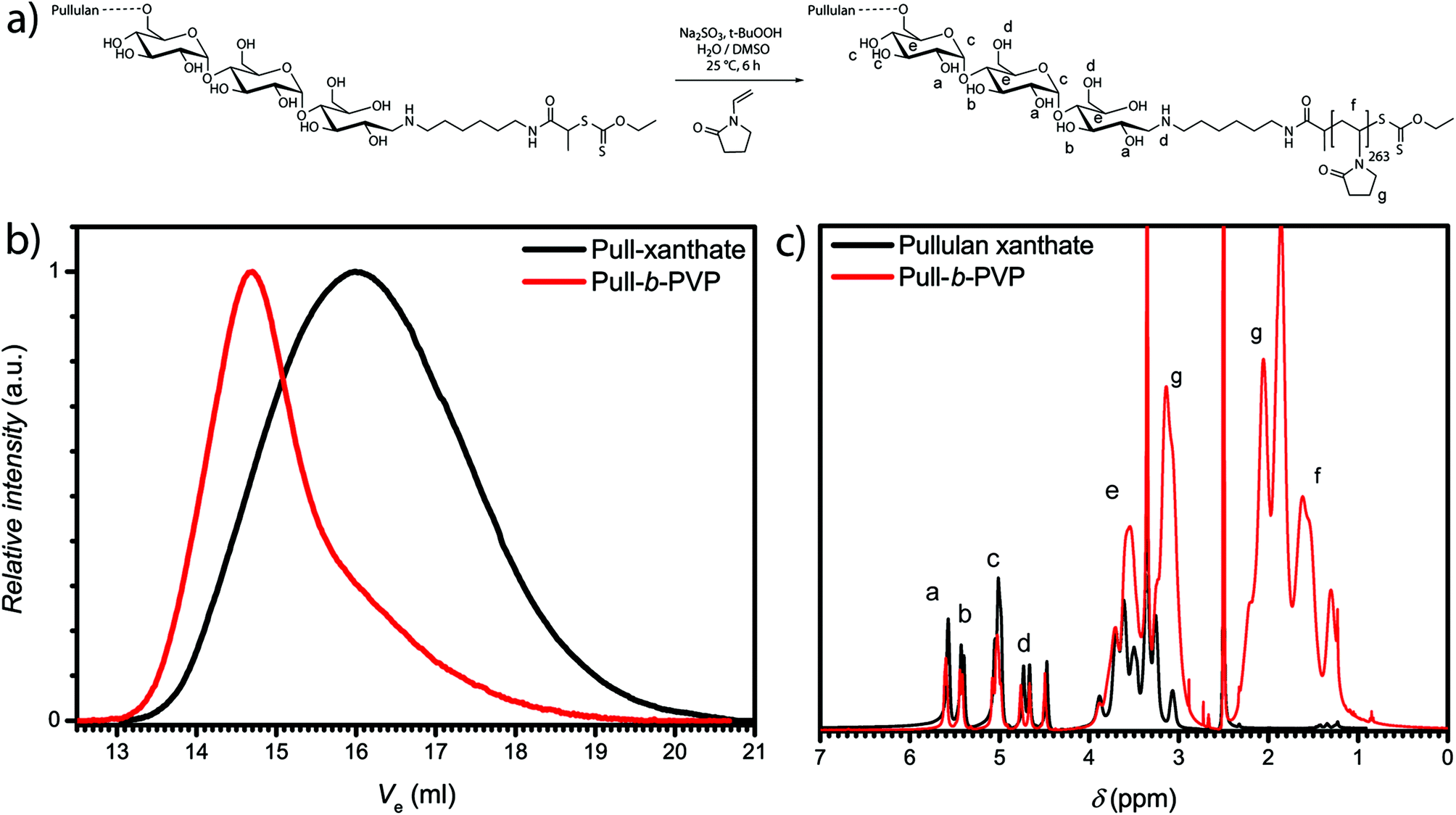 | ||
| Fig. 1 (a) Polymerization scheme for Pull-b-PVP block copolymer formation in water/DMSO mixture, (b) SEC traces of pullulan xanthate and Pull124-b-PVP263 block copolymer synthesized in water/DMSO mixtures recorded in NMP at 70 °C and (c) 1H-NMR of pullulan xanthate and Pull124-b-PVP263 recorded at 400 MHz in DMSO-d6. | ||
Nevertheless, the SEC traces (Fig. 1b) display slight tailing, which can be explained with the broad molecular weight distribution of pullulan. Additionally, increased interactions with the SEC column as well as the occurrence of dead chains during the polymerization can play a role for the observed tailing. The 1H-NMR spectrum (Fig. 1c) further displays the successful block copolymerization with the proton signals of both polymer blocks between 4.4 ppm and 5.7 ppm for pullulan and from 1.2 ppm to 2.1 ppm for PVP, being present in the spectrum. According to the integration of the protons corresponding to the a-OH group of pullulan (Fig. 1c) and the relation to the integral of the protons corresponding to the PVP backbone, indexed as f (Fig. 1c) in the 1H-NMR spectrum, a VP incorporation of 263 repeating units was calculated. The block copolymer composition was therefore assessed to be Pull124-b-PVP263. In the next step the self-assembly behavior of Pull-b-PVP was investigated in diluted aqueous solutions.
Self-assembly of Pull-b-PVP block copolymers in aqueous solution
After the successful synthesis of Pull124-b-PVP263, the self-assembly behavior was investigated. Therefore, solutions containing 0.1, 0.5, 1.0, and 2.5 wt% of Pull124-b-PVP263 in Millipore water were prepared, filtered with 0.45 μm CA syringe filters, and analyzed via DLS. In order to compare the block copolymer with the pullulan homopolymer, a pullulan solution containing 0.1 wt% in Millipore water was prepared as well and investigated via DLS (Table 1).| Polymer | Concentration (wt%) | Peak 1 Rh,app (nm) | Rel. abund. | Peak 2 Rh,app (nm) | Rel. abund. |
|---|---|---|---|---|---|
| Pullulan | 0.1 | 7 | 1.0 | 180 | 0.7 |
| Pull124-b-PVP263 | 0.1 | 25 | 1.0 | — | — |
| 0.5 | 16 | 1.0 | 750 | 0.12 | |
| 1.0 | 11 | 1.0 | 350 | 0.66 | |
| 2.5 | 7 | 0.42 | 800 | 1.0 |
The intensity weighted size distributions of pullulan and Pull124-b-PVP263 at 0.1 wt% in Fig. 2a (Fig. S8†) display a strong difference. Pullulan shows a bimodal particle size distribution with the first peak being the most abundant species with an average apparent radius of 7 nm. The peak can be attributed to free dissolved pullulan macromolecules. The second peak possesses an average apparent radius of 180 nm with a relative abundance of 0.7, which corresponds to weak aggregates of pullulan chains. The aggregates presumably form because of hydrogen bonding between hydroxyl groups and entanglement of the pullulan chains. Due to the intensity weighted particle size distribution, the actual abundance of these aggregates is 10−5 times lower compared to the free dissolved polymer. In contrast to free pullulan, the block copolymer only displays a unimodal particle size distribution with an average apparent radius of 25 nm. This is a further indication of the successful block copolymer formation as the self-assembly behavior in solution is significantly different from the pullulan homopolymer. Despite this fact, the self-assembly of Pull124-b-PVP263 block copolymers is not as efficient as expected for low concentrations. Instead of large structures that would indicate the presence of particles or vesicular structures, only one signal corresponding either to free dissolved block copolymer or small aggregates could be observed. Since the average apparent radius is with 25 nm quite large for free dissolved block copolymer chains, the presence of small aggregates of a low amount of block copolymer chains is more conceivable.
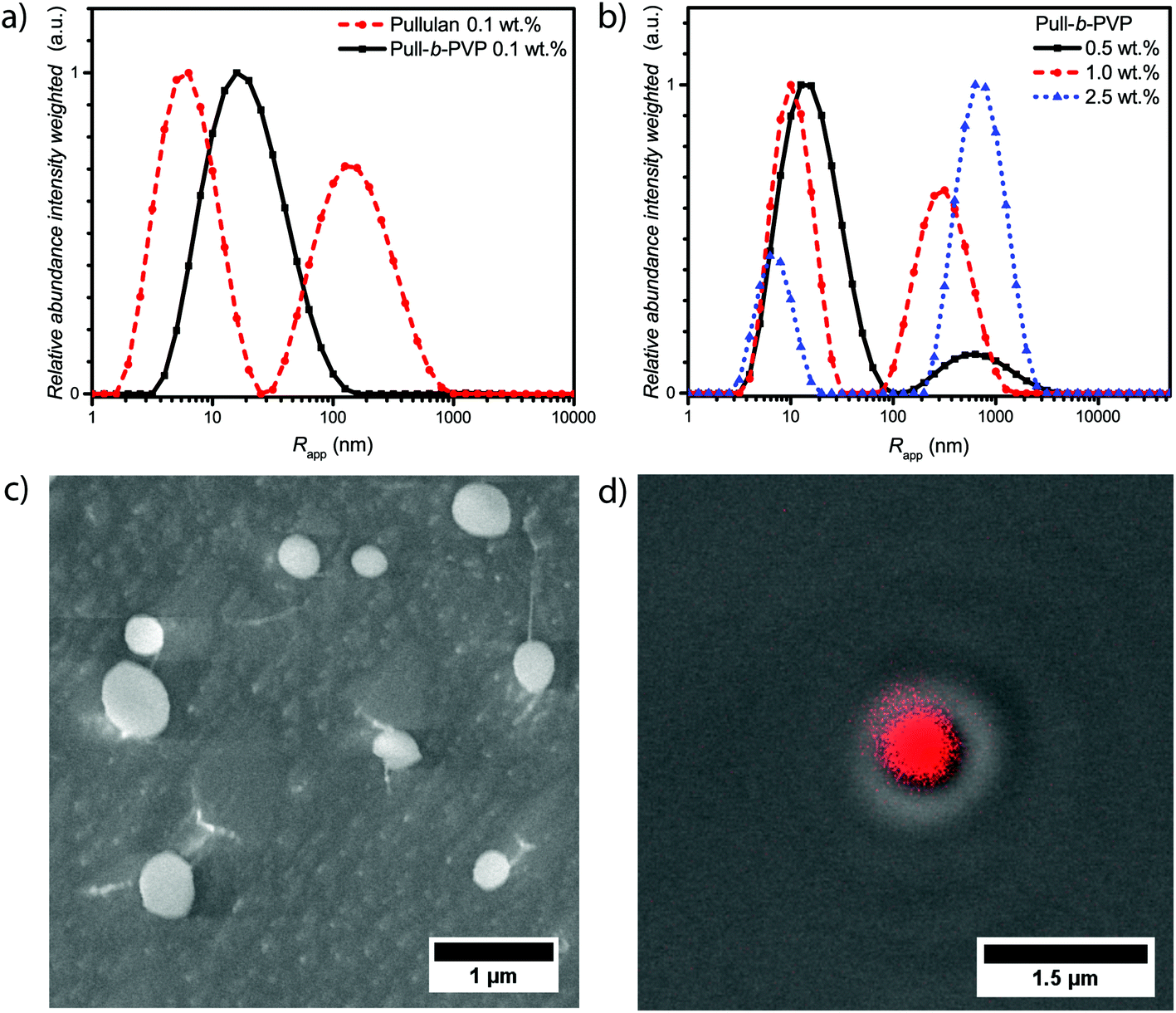 | ||
| Fig. 2 (a) Intensity weighted size distributions of pullulan and Pull-b-PVP at 0.1 wt%; (b) intensity weighted size distributions of Pull-b-PVP at higher concentrations in Millipore water measured via DLS at 25 °C; (c) cryo SEM micrographs of a 0.5 wt% Pull124-b-PVP263 solution and (d) differential interference contrast (DIC) image with confocal laser scanning microscopy (CLSM) overlay displaying the particle structure of Pull-b-PVP at 2.5 wt% stained with 0.08 mM Rhodamine B. | ||
An increase in concentration to 0.5 wt% already has a decent effect on the self-assembly behavior of the block copolymer (Fig. 2b and Fig. S8†). In contrast to the 0.1 wt% sample, a bimodal particle size distribution is present with a second peak with an apparent average radius of 750 nm and a relative abundance of 0.12. The decrease in the average apparent radius of the small species and the formation of very large aggregates indicates a concentration dependent tendency to form larger structures. A similar behavior was already observed for PEO-b-PVP block copolymers, however with a lower tendency of self-assembly.33 An increase in the block copolymer concentration to 1.0 wt% and 2.5 wt% further decreases the average apparent radius and the abundance of particle species increases. With an average apparent radius of the large species of 800 nm at 2.5 wt%.
The cryo SEM measurements of a 0.5 wt% solution of Pull124-b-PVP263 display an increased amount of spherical particles with average diameters between 150 nm and almost 1 μm (Fig. 2c). Moreover, tubular structures containing free dissolved block copolymer can be observed (Fig. S9†). The tubular structures form due to the growth of ice crystals during the freezing process in liquid nitrogen pushing the free dissolved block copolymers to the crystal border, where they concentrate and form tubular alignments.48 Furthermore, string-like connections between a significant amount of smaller particles can be observed, e.g. the particles are aligned in the fashion of pearls that are lined up on a string composed of block copolymer (Fig. S9†). The occurrence of these interparticle connections is not completely understood yet and is possibly an artifact of the cryo SEM process. Nonetheless, a high amount of spherical particles could be confirmed via cryo SEM measurements. In agreement to the intensity weighed particle size distribution of the 0.5 wt% solution from DLS, the amount of free dissolved block copolymer is significantly present in the cryo SEM sample as tubular shaped structures.
Subsequently CLSM measurements were carried out with a Rhodamine B stained 2.5 wt% solution of Pull124-b-PVP263 in order to support the structure formation postulated by DLS and cryo SEM. Therefore, the polymer solution was stained with a 0.08 mM Rhodamine B solution and investigated with CLSM techniques. Spherical particles with a diameter between 600 nm and 1 μm could be observed in the confocal micrographs (Fig. 2d and S10†). Moreover, the DIC micrographs with fluorescence overlay show that the particles are highly enriched with Rhodamine B (Fig. 2d and S10†). In addition to observed single particles, agglomerates of several particles were observed as well (Fig. S10†). Since the structures were imaged directly at the surface of the glass slide in order to decrease blurring effects due to increased particle motion, the occurrence of the particle agglomerates can be explained by a continuous sinking of particles from the solution onto the same area of the glass surface. The highly increased amount of dye inside the spherical structures compared to the surrounding solution can be attributed to the same phenomenon as recently described for Pull-b-PDMA and Pull-b-PEA block copolymer solutions.21 Rhodamine B can permeate through the membrane structure inside the self-assembled particles, but remains inside the structure due to an increased interaction between dye molecules and block copolymer.
A comparison of the obtained average diameters from CLSM/DIC and cryo SEM with the average apparent radii obtained from DLS shows significant differences at the corresponding concentrations. The average apparent diameters calculated from DLS exceed the diameters determined via microscopy about a factor of 30–50%, which might be due to the intensity weighted particle size distribution. As larger particle are overexpressed in contrast to smaller ones in the distribution curve. Nevertheless, particle formation of completely hydrophilic block copolymers in aqueous solution is evident. In order to stabilize the particles against dilution and open up opportunities for future applications, crosslinking of the particles was studied subsequently.
Oxidation of Pull-b-PVP and crosslinking of Pull-b-PVP vesicles
The confirmation of spherical particles self-assembled from Pull-b-PVP block copolymers already displays the potential of this system. In order to preserve the structures and generate more applicable systems for future utilization in drug delivery, a crosslinking strategy has to be developed. Since the application of polysaccharides in biomedical and pharmaceutical field as hydrogel components is well studied, several techniques to crosslink polysaccharides with or without the application of reversible crosslinking agents were reported.47,49–52 The reversibility of the crosslinking was usually enabled via crosslinkers bearing redox responsive groups such as dithiols or a pH sensitive linkage with the polysaccharide, such as imines.51 To enable imine formation with polysaccharides such as dextran or pullulan, the cyclic hexoses need to be oxidized to aldehydes. The most frequently reported pathway to accomplish a mild oxidation of polysaccharides without the destruction of the backbone is carried out with sodium (meta)periodate in aqueous solutions (see ESI† for details).49,50 Maia et al. reported a facile oxidation and crosslinking method for dextran.47 In order to investigate the possibility of selective pullulan oxidation to a pullulan dialdehyde, test oxidations with depolymerized pullulan with different percentages of NaIO4 as oxidizing agent were conducted (Table S2†). The state of oxidation was characterized via1H-NMR spectroscopy (Fig. S11 and Table S3†). Furthermore, SEC measurements should indicate depolymerization or degradation of oxidized pullulan (Fig. S12†). In addition to the formation of aldehyde units, a decrease in pullulan molecular weight was observed for high NaIO4 equivalents that might be due to fractured polymer chains. For that reason, oxidations exceeding 10% of NaIO4 were not conducted with the Pull124-b-PVP263 block copolymers.In order to oxidize the pullulan moieties of a Pull-b-PVP block copolymer, the DHBC was dissolved in Millipore water and the corresponding amount of NaIO4 was added to oxidize 5% and 10% of the pullulan units to dialdehyde (Fig. 3a). As visible from the 1H-NMR spectra in Fig. 3b, the oxidation of glucose units of Pull124-b-PVP263 can be regarded as successful. The appearance of the anomeric aldehyde protons in the area between 6.1 ppm and 7.5 ppm confirms a successful oxidation procedure. The integrals normalized to the internal standard DMF display an increase from the control sample with an integral of 0.04 to 0.67 and 1.35 for the 5% and 10% oxidation, respectively. However, absolute values of oxidized units cannot be determined via the previous method because the exact molecular mass of the block copolymer is unknown and only the apparent average number weighted molecular masses were determined. Therefore, no clear statement on the actual oxidation state can be given, but from the comparison of the integrals corresponding to 5% oxidation and 10% oxidation a two fold increase in the absolute value of anomeric aldehyde protons can be stated.
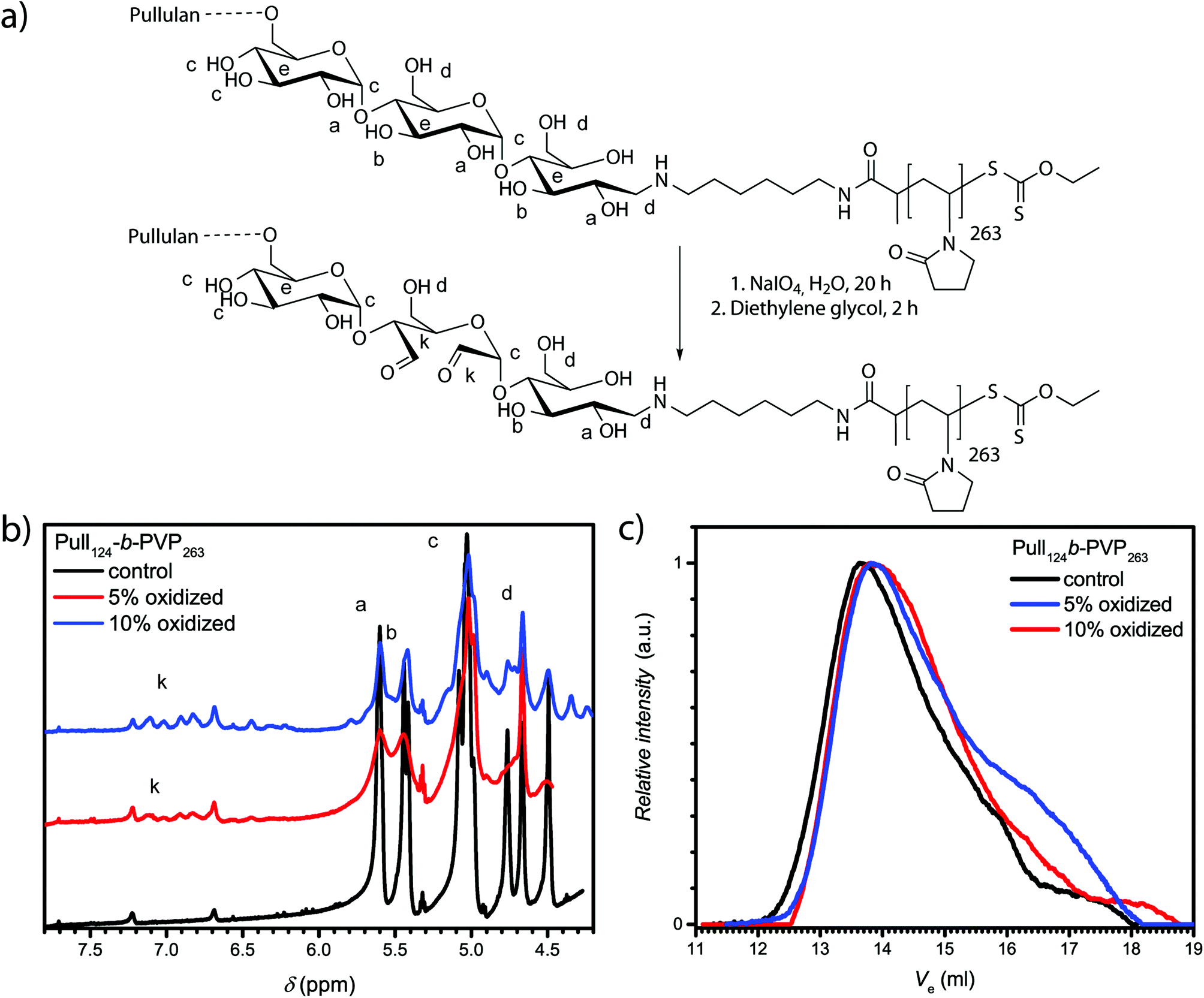 | ||
| Fig. 3 (a) Schematic oxidation of Pull124-b-PVP263 with NaIO4; (b) 1H-NMR spectra of oxidized Pull124-b-PVP263 emphasizing on the anomeric proton peaks of the oxidized glucose units recorded at 400 MHz in DMSO-d6; (c) corresponding SEC elution curves recorded in NMP at 70 °C. | ||
The SEC elution curves in Fig. 3c display a decent change of the elution curves upon oxidation with NaIO4. The oxidized block copolymers elute with a significantly broader curve compared to the initial block copolymer. The drastic change in the appearance of the elugrams can be attributed to a different interaction of the oxidized block copolymer with the solvent NMP resulting in a peak broadening towards higher elution volumes and a slightly more pronounced tailing. Another reason for tailing towards longer retention times might be chain degradation after the oxidation as species of lower molecular weight are present. Nevertheless, polymer chain fracture or depolymerization occurs only to a minor extent as seen from the elugrams and a successful oxidation of pullulan is indicated. Therefore, the appearance of the anomeric proton signals in the 1H-NMR spectrum and the SEC results both point towards a successful oxidation.
As already indicated by SEC measurements a change in the chemical behavior of the block copolymer is induced by oxidation of the Pull backbone, which is also indicated by DLS measurements of aqueous solutions of Pull124-b-PVP263 and its oxidized derivatives at 0.1 wt%. As visible from the particle size distribution in Fig. 4 the unimodal average apparent particle size distribution of Pull-b-PVP at 0.1 wt% turned to a bimodal distribution for both oxidized species. The apparent average particle size distributions of the oxidized block copolymers show an increased tendency for self-assembly already at lower concentrations in contrast to non-oxidized Pull-b-PVP. The origin of the increased tendency can be referred to a change in the hydrophilicity of pullulan. As oxidation of alcohols to aldehydes decreases hydrophilicity and increases the probability of hydrogen bonding of pullulan, microphase separation is encouraged. An enhanced self-assembly is very beneficial for the attempted crosslinking step. As shown for higher concentrated solutions of Pull-b-PVP (Fig. 2b), the tendency to form structures such as particles increases with raising concentration. Conclusively, a higher amount of self-assembled structures should be present for the oxidized species at more concentrated solutions than it was the case for general Pull-b-PVP.
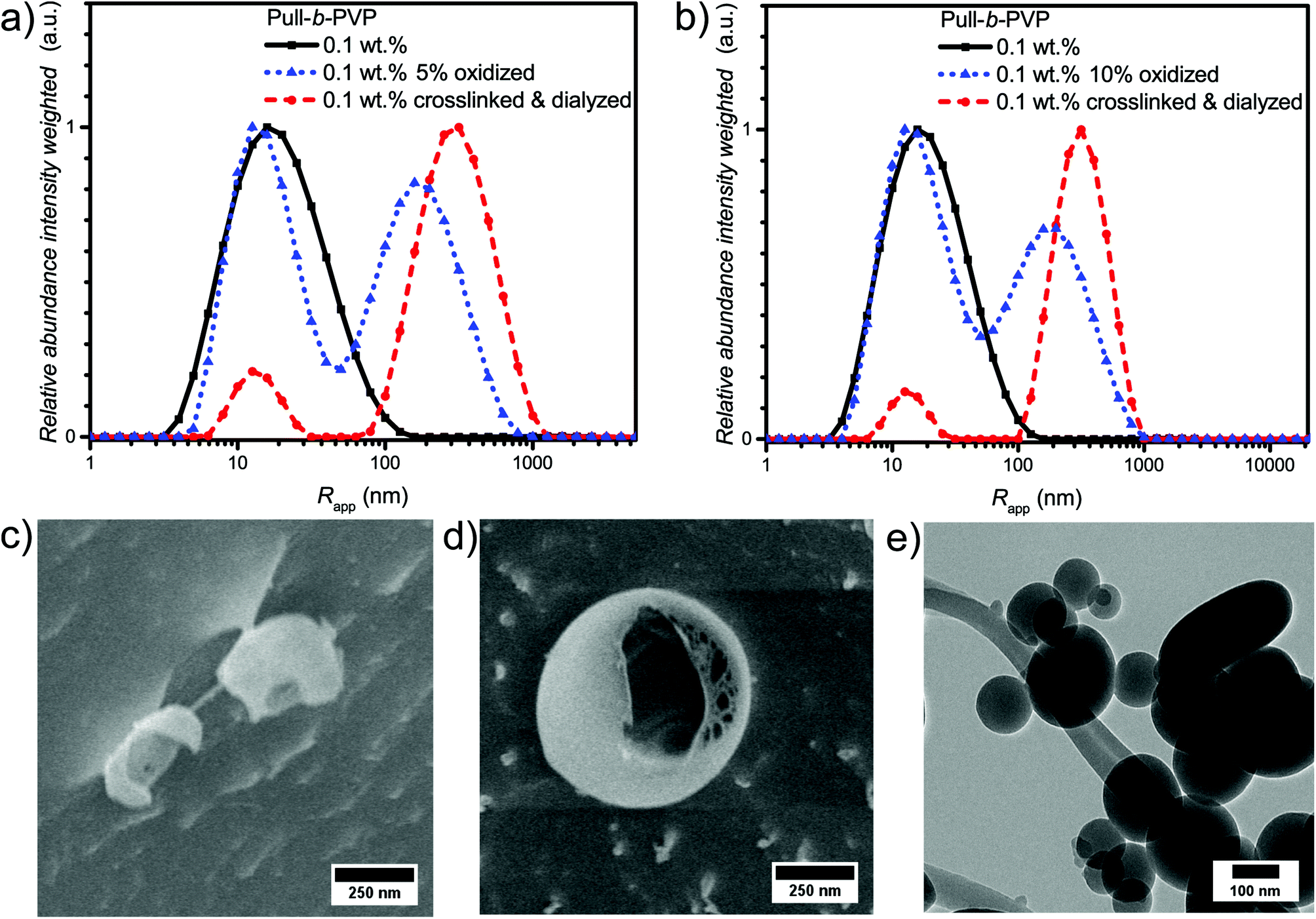 | ||
| Fig. 4 Intensity weighted particle size distributions of 0.1 wt% solutions of initial Pull124-b-PVP263, oxidized block copolymer before and after crosslinking and dialysis with (a) 5% of oxidized pullulan and (b) 10% of oxidized pullulan in Millipore water measured via DLS at 25 °C. Cryo SEM micrographs of Pull124-b-PVP263 particles at a concentration of 0.1 wt% after crosslinking and dialysis with (c) 5% of oxidized pullulan and (d) 10% of oxidized pullulan. (e) TEM image of 10% oxidized Pull124-b-PVP263 after crosslinking and freeze drying on a copper grid. | ||
Due to the enhanced amount of self-assembled structures at higher block copolymer concentrations, the crosslinking procedure was conducted at a highly concentrated state of 5.0 wt%. Cystamine dihydrochloride was selected as crosslinking agent. The crosslinker can reversibly attach to the oxidized glucose units of the pullulan block via imine formation with aldehyde groups. Furthermore, the disulfide bridge can be cleaved redox chemically by the application of suitable reducing agents, such as tricarboxyethyl phosphine (TCEP). Thus, two different triggers, pH and redox, can be applied for disassembly and probably release applications. To conduct the crosslinking, an aqueous solution containing 5 wt% of Pull124-b-PVP263 was prepared and a 0.044 mmol mL−1 cystamine dihydrochloride solution was added. The mixture was shaken for 36 hours in order to achieve a high crosslinking density (Scheme 2). The DLS distributions do not show significant differences before and after crosslinking (Fig. S13†). In order to remove free dissolved block copolymer as well as dimers and lower aggregates, the crosslinked solution was diluted to 0.1 wt% and dialyzed for three days with an 1000000 MWCO dialysis tube against Millipore water. The high MWCO of the dialysis membrane and the long dialysis time should ensure a complete removal of smaller species, but trap the larger crosslinked aggregates inside the tube.
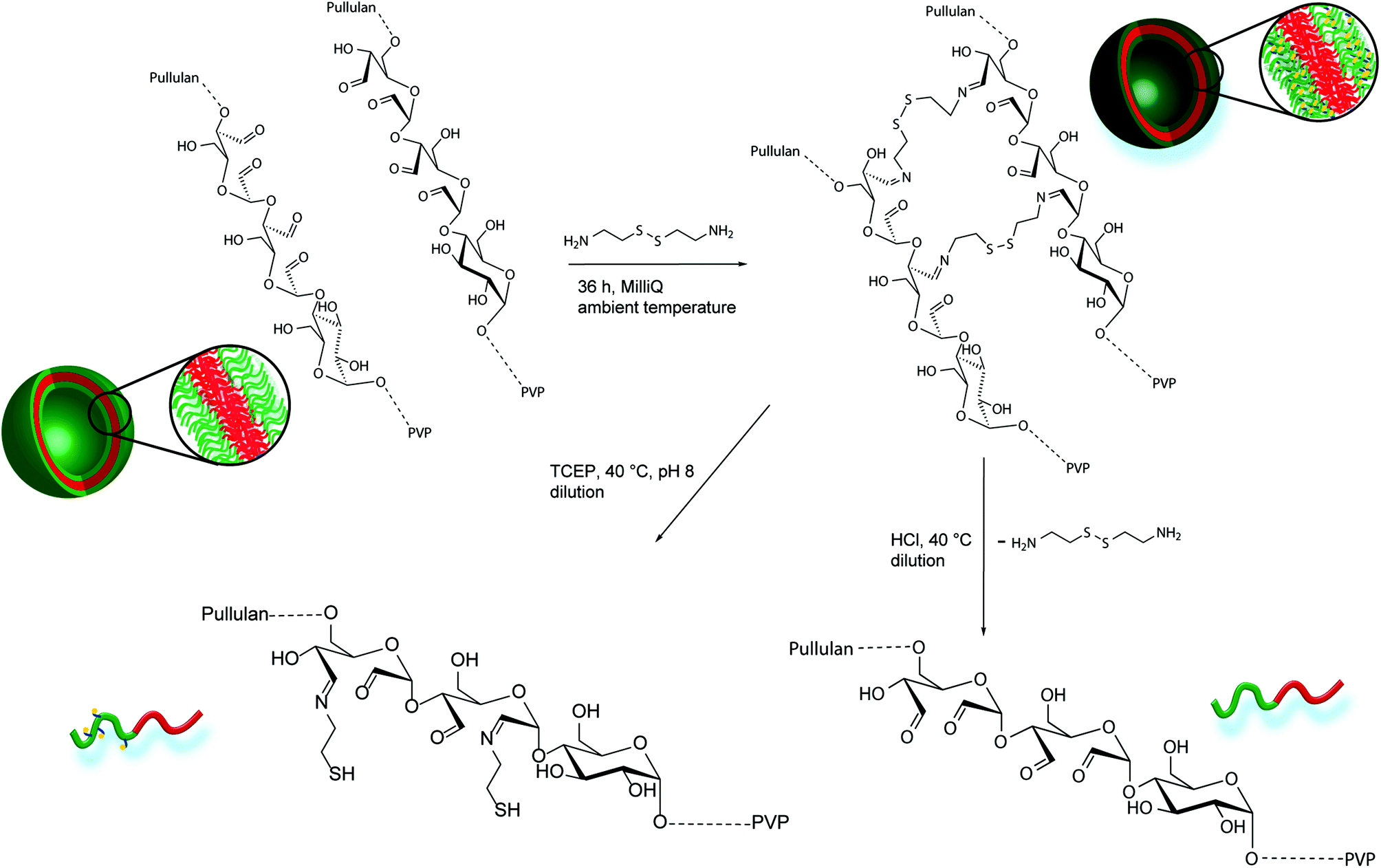 | ||
| Scheme 2 Schematic crosslinking procedure of oxidized Pull-b-PVP self-assemblies with cystamine in Millipore water, pH or redox induced disassembly of crosslinked Pull-b-PVP vesicles. | ||
As visible from the intensity weighted particle size distributions of the oxidized Pull-b-PVP block copolymer before, after crosslinking and dialysis (Fig. 4a), crosslinking and dialysis drastically shifted the average apparent particle size distribution towards the large species. At the same concentration of 0.1 wt% the amount of unimers would be significantly increased without crosslinking. Thus, crosslinking preserves larger particles under diluted conditions. The relative abundance of the free dissolved 5% oxidized block copolymer species decreased by 80% from 1.0 to 0.2 while maintaining the average apparent radius of 14 nm. Nevertheless, complete removal of unimer species was not possible via dialysis. Moreover, the average apparent radius of the large particle species increased to 340 nm being the most abundant species by intensity weighting. However, free dissolved block copolymer is still present in the solution. Due to the intensity weighted size distribution, the abundance of the free dissolved block copolymer species appears to be significantly lower than it is the case in reality. Therefore, it can be stated that dialysis was not able to completely remove all free dissolved species. One explanation for this inability could be the reversibility of the imine formation due to concentration and entropic reasons leading to slight disassembly with time which may result in free dissolved block copolymer still being abundant in the solution of crosslinked structures. The crosslinked and dialyzed sample of Pull124-b-PVP263 oxidized to 10% displays a similar bimodal intensity weighted particle size distribution with average apparent radii of 14 nm for the free dissolved block copolymer and 340 nm for the crosslinked structures (Fig. 4b and Table S4†). Again, complete removal of small species was not possible via dialysis.
In addition, the cryo SEM micrographs of the crosslinked and dialyzed sample of Pull124-b-PVP263 oxidized to 5% display spherical particles and a small amount of vesicular structures (Fig. 4c and S14†). Moreover, no tubular alignments of free dissolved block copolymer are visible as before (Fig. S14†). The crosslinked and dialyzed particles possess average diameters between 250 nm and 1.2 μm. Larger spherical structures with diameters between 700 nm and 1.2 μm often show a ruptured morphology with a hollow interior indicating vesicle formation. Moreover, some ruptured smaller particles with diameters around 250 nm could be observed (Fig. S14†) which indicate a hollow structure as well. The presence of vesicular structures increases with a higher oxidation state of pullulan at 10%. The cryo SEM micrographs (Fig. 4d and S15†) display a high amount of vesicular structures with average diameters ranging between 250 nm and 1.2 μm. Only a small amount of small structures without vesicular shape could be observed (Fig. S15†). An even more impressive observation can be seen in Fig. S15† with the presence of anisotropic structures, which could possibly origin from two structures that merged during the crosslinking process. Such a merging of vesicles is not unlikely due to the high concentration of 5.0 wt% during the crosslinking procedure, which results in close proximity of the spherical structures and entanglement of block copolymer chain is possible. The magnification of a crosslinked Pull-b-PVP vesicle (Fig. 4d) gives some more insight into its interior displaying an interpenetrating network of crosslinked block copolymer which increases in density towards the outer border to form a closed shell. Despite the insight into the vesicular structure, the cryo SEM technique prevents a clear postulation about the origin of the observations. These network structures can either be an intrinsic feature of the vesicles or be artifacts of the sample preparation, especially during the freezing and sputtering process.
TEM imaging of 10% oxidized Pull124-b-PVP263 after crosslinking and freeze drying on a copper grid (Fig. 4e and S16†) shows spherical particles with diameters between 100 and 500 nm. The particles might appear smaller than expected as drying of the samples certainly leads to collapsed structures. Moreover, some particles seem to have ruptured shells (Fig. S16b†), which is another indication for vesicular structures being present in solution.
Despite the uncertainties regarding the interior vesicle structure, the self-assembled and crosslinked vesicles already mark a significant step towards a model drug delivery cargo system. The vesicular structures encourage an encapsulation of target molecules. Therefore, a triggered cleavage of the crosslinked structures would be beneficial, e.g. via a pH or redox trigger (Scheme 2).
In order to assess pH triggered disassembly of the self-assembled and crosslinked Pull-b-PVP vesicles, 2.0 mL of 0.1 wt% solutions of the 5% oxidized and crosslinked samples were treated with HCl. The pH of the solution was adjusted to 3 (150 μL of a 0.1 mol L−1 HCl solution) and the solution was heated to 40 °C overnight. Reduction triggered disassembly was performed via TCEP addition to self-assembled and crosslinked Pull-b-PVP vesicles, 2.0 mL of 0.1 wt% solutions of the 10% oxidized and crosslinked samples were treated with 20 mg TCEP for the cleavage of the disulfide bridges. Therefore, the solution containing the crosslinked vesicles was placed in a DLS vial and argon was bubbled through to remove the oxygen inside the solution. Subsequently, TCEP was added and the sealed vial was heated in a water bath at 40 °C overnight. In both cases heat treatment was necessary to generate a certain activation energy which is required to accelerate the desired reaction pathway and encourage the disassembly process. After the addition of the cleavage agent and heat treatment, the two samples were investigated via DLS and compared with the particle size distribution of the oxidized and the crosslinked samples (Fig. 5).
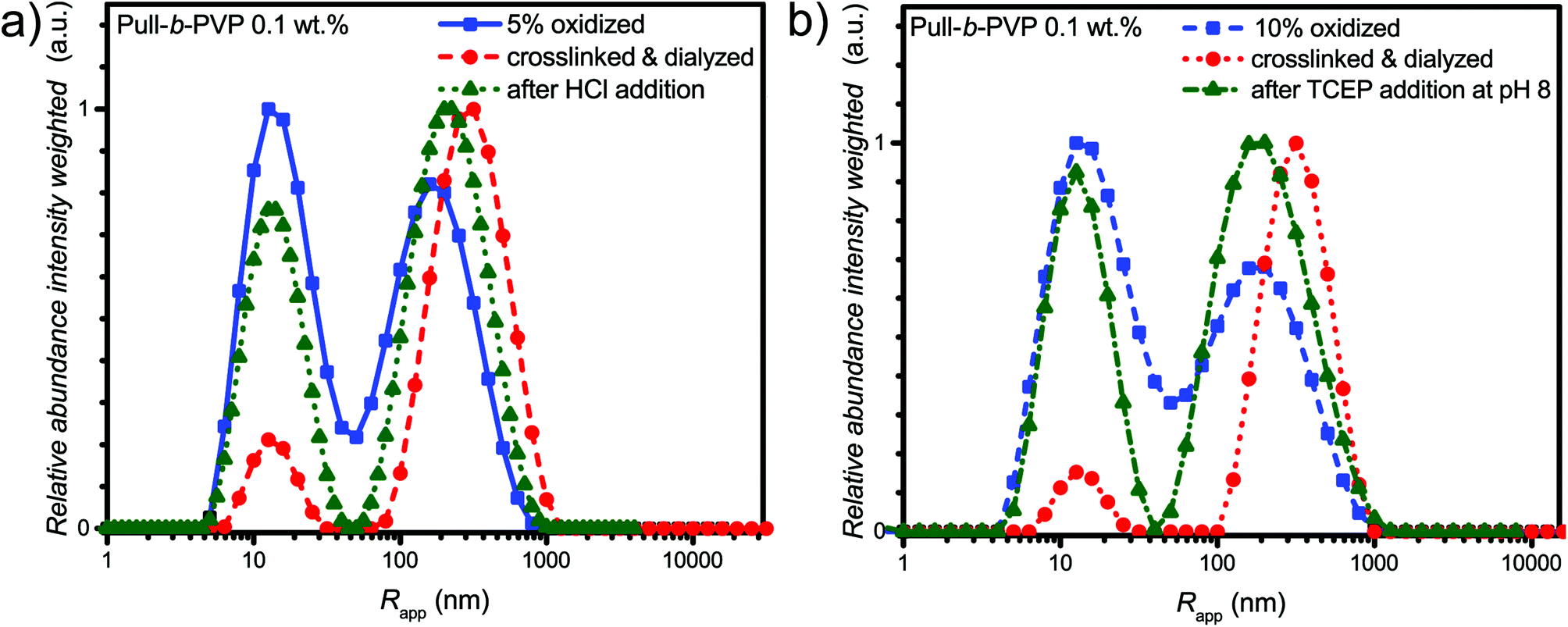 | ||
| Fig. 5 Comparison of intensity weighted particle size distributions of 0.1 wt% solutions in Millipore water measured via DLS at 25 °C of (a) 5% oxidized Pull124-b-PVP263, oxidized block copolymer after crosslinking and dialysis and after treatment with HCl; (b) 10% oxidized Pull124-b-PVP263, oxidized block copolymer after crosslinking and dialysis and after treatment with TCEP. | ||
The DLS results of the acid treatment of the crosslinked Pull-b-PVP vesicles (Fig. 5a) display a significant disassembly of the crosslinked structures. The relative abundance of the free dissolved species increased by four-fold to 0.77 and the average apparent radius decreased by 27% from 340 nm to 250 nm (Table S4†). Nevertheless, complete disassembly was not observed but the observed distribution is close to the initial distribution after oxidation. Moreover, in the case of TCEP treatment as visible from the intensity weighted apparent average particle size distribution (Fig. 5b) the addition of TCEP to the crosslinked vesicle solution resulted in disassembly of the vesicular structures as well. The relative abundance of the peak corresponding to the free dissolved block copolymer increased by four fold from 0.2 to 0.92. Taking the intensity weighing into account, the increase in relative abundance is already very significant and a decent success in disassembly can be stated. Nevertheless, quantitative disassembly was not the case, yet the apparent size distribution is close to the initial size distribution after oxidation. Furthermore, the average apparent radius decreased by 30% from 340 nm to 240 nm (Table S4†). When comparing the average apparent particle size distribution of the oxidized species with the crosslinked one after TCEP addition, a complete disappearance of the aggregated species can probably not be achieved because the oxidized species displays a certain tendency to form aggregates as well. Overall, acid and TCEP addition display a facile pathway towards the disassembly of the crosslinked DHBC vesicles.
Conclusions
Herein, a novel DHBC Pull-b-PVP was synthesized via RAFT/MADIX techniques starting from the biomacromolecule pullulan and the related macro RAFT/MADIX chain transfer agent. The block copolymer self-assembled to spherical structures with an average apparent radius of 800 nm at increased concentrations in water. Furthermore, spherical structures could be observed with cryo SEM and CLSM techniques. The pullulan block could be successfully converted to present aldehyde groups acting as anchor point for crosslinker attachments. It was demonstrated, that the oxidized self-assembled particles could be crosslinked via the bifunctional crosslinker cystamine forming dynamic covalent imine linkages with aldehyde groups. The afforded vesicles with an average diameter of 700 nm were stable upon high dilution and could be observed via cryo SEM and TEM. Furthermore, it was possible to cleave the crosslinking bonds with the treatment of acid or the application of the reducing agent TCEP. The triggered cleavage is a key feature taking future applications in the biomedical sector into account.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful for funding from the Max-Planck society. The authors would like to acknowledge Marlies Gräwert for SEC measurements, Heike Runge for assistance with cryogenic SEM and TEM measurements, Dr. Tom Robinson as well as Carmen Remde for assistance with CLSM measurements. We thank Prof. Markus Antonietti for fruitful discussions. Open Access funding provided by the Max Planck Society.Notes and references
- C. J. Hawker and K. L. Wooley, Science, 2005, 309, 1200–1205 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- T. H. Epps III and R. K. O'Reilly, Chem. Sci., 2016, 7, 1674–1689 RSC.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS PubMed.
- E. Krämer, S. Förster, C. Göltner and M. Antonietti, Langmuir, 1998, 14, 2027–2031 CrossRef.
- M. C. Orilall and U. Wiesner, Chem. Soc. Rev., 2011, 40, 520–535 RSC.
- J. Y. Cheng, A. M. Mayes and C. A. Ross, Nat. Mater., 2004, 3, 823–828 CrossRef CAS PubMed.
- M. Inam, G. Cambridge, A. Pitto-Barry, Z. P. L. Laker, N. R. Wilson, R. T. Mathers, A. P. Dove and R. K. O'Reilly, Chem. Sci., 2017, 8, 4223–4230 RSC.
- G. Riess, Prog. Polym. Sci., 2003, 28, 1107–1170 CrossRef CAS.
- J.-F. Gohy, in Block Copolymers II, ed. V. Abetz, Springer, Berlin, Heidelberg, 2005, pp. 65–136 Search PubMed.
- E. Blasco, B. V. K. J. Schmidt, C. Barner-Kowollik, M. Pinol and L. Oriol, Polym. Chem., 2013, 4, 4506–4514 RSC.
- N. Petzetakis, A. P. Dove and R. K. O'Reilly, Chem. Sci., 2011, 2, 955–960 RSC.
- J. B. Gilroy, T. Gädt, G. R. Whittell, L. Chabanne, J. M. Mitchels, R. M. Richardson, M. A. Winnik and I. Manners, Nat. Chem., 2010, 2, 566–570 CrossRef CAS PubMed.
- B. Jeong, Y. H. Bae, D. S. Lee and S. W. Kim, Nature, 1997, 388, 860–862 CrossRef CAS PubMed.
- A. Rösler, G. W. M. Vandermeulen and H.-A. Klok, Adv. Drug Delivery Rev., 2012, 64, 270–279 CrossRef.
- S. Förster and M. Antonietti, Adv. Mater., 1998, 10, 195–217 CrossRef.
- S.-J. Park, S.-G. Kang, M. Fryd, J. G. Saven and S.-J. Park, J. Am. Chem. Soc., 2010, 132, 9931–9933 CrossRef CAS PubMed.
- C. Hörenz, C. Pietsch, A. S. Goldmann, C. Barner-Kowollik and F. H. Schacher, Adv. Mater. Interfaces, 2015, 2, 1500042 CrossRef.
- B. V. K. J. Schmidt and C. Barner-Kowollik, Polym. Chem., 2014, 5, 2461–2472 RSC.
- S. M. Brosnan, H. Schlaad and M. Antonietti, Angew. Chem., Int. Ed., 2015, 54, 9715–9718 CrossRef CAS PubMed.
- J. Willersinn, A. Bogomolova, M. Brunet Cabré and B. V. K. J. Schmidt, Polym. Chem., 2017, 8, 1244–1254 RSC.
- J. Willersinn and B. V. K. J. Schmidt, Polymers, 2017, 9, 293 CrossRef.
- H. Park, S. Walta, R. R. Rosencrantz, A. Korner, C. Schulte, L. Elling, W. Richtering and A. Boker, Polym. Chem., 2016, 7, 878–886 RSC.
- A. Taubert, E. Furrer and W. Meier, Chem. Commun., 2004, 2170–2171 RSC.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed.
- A. B. Lowe and C. L. McCormick, Prog. Polym. Sci., 2007, 32, 283–351 CrossRef CAS.
- J. F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS PubMed.
- J. Wu, Z. Wang, Y. Yin, R. Jiang, B. Li and A.-C. Shi, Macromolecules, 2015, 48, 8897–8906 CrossRef CAS.
- A. F. Hirschbiel, B. V. K. J. Schmidt, P. Krolla-Sidenstein, J. P. Blinco and C. Barner-Kowollik, Macromolecules, 2015, 48, 4410–4420 CrossRef CAS.
- W. Zhang, Z. Kochovski, B. V. K. J. Schmidt, M. Antonietti and J. Yuan, Polymer, 2016, 107, 509–516 CrossRef CAS.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59–63 CrossRef CAS PubMed.
- B. Kumru, M. Shalom, M. Antonietti and B. V. K. J. Schmidt, Macromolecules, 2017, 50, 1862–1869 CrossRef CAS.
- J. Willersinn, M. Drechsler, M. Antonietti and B. V. K. J. Schmidt, Macromolecules, 2016, 49, 5331–5341 CrossRef CAS.
- V. Ďorďovič, M. Uchman, K. Procházka, A. Zhigunov, J. Pleštil, A. Nykänen, J. Ruokolainen and P. Matějíček, Macromolecules, 2013, 46, 6881–6890 CrossRef.
- R. S. Singh, N. Kaur and J. F. Kennedy, Carbohydr. Polym., 2015, 123, 190–207 CrossRef CAS PubMed.
- L. Chen, X. Wang, F. Ji, Y. Bao, J. Wang, X. Wang, L. Guo and Y. Li, RSC Adv., 2015, 5, 94719–94731 RSC.
- C. Schatz, S. Louguet, J.-F. Le Meins and S. Lecommandoux, Angew. Chem., Int. Ed., 2009, 48, 2572–2575 CrossRef CAS PubMed.
- M. Teodorescu and M. Bercea, Polym.-Plast. Technol. Eng., 2015, 54, 923–943 CrossRef CAS.
- C. R. Mace, O. Akbulut, A. A. Kumar, N. D. Shapiro, R. Derda, M. R. Patton and G. M. Whitesides, J. Am. Chem. Soc., 2012, 134, 9094–9097 CrossRef CAS PubMed.
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- Y. Azuma, T. Terashima and M. Sawamoto, Macromolecules, 2017, 50, 587–596 CrossRef CAS.
- J. Xu, K. Jung, N. A. Corrigan and C. Boyer, Chem. Sci., 2014, 5, 3568–3575 RSC.
- W. C. de Vries, D. Grill, M. Tesch, A. Ricker, H. Nüsse, J. Klingauf, A. Studer, V. Gerke and B. J. Ravoo, Angew. Chem., Int. Ed., 2017, 56, 9603–9607 CrossRef CAS PubMed.
- F. Lecolley, L. Tao, G. Mantovani, I. Durkin, S. Lautru and D. M. Haddleton, Chem. Commun., 2004, 2026–2027 RSC.
- L. Ilić, K. Jeremić and S. Jovanović, Eur. Polym. J., 1991, 27, 1227–1229 CrossRef.
- G. Pound, F. Aguesse, J. B. McLeary, R. F. M. Lange and B. Klumperman, Macromolecules, 2007, 40, 8861–8871 CrossRef CAS.
- J. Maia, L. Ferreira, R. Carvalho, M. A. Ramos and M. H. Gil, Polymer, 2005, 46, 9604–9614 CrossRef CAS.
- B. Wicklein, A. Kocjan, G. Salazar-Alvarez, F. Carosio, G. Camino, M. Antonietti and L. Bergstrom, Nat. Nanotechnol., 2015, 10, 277–283 CrossRef CAS PubMed.
- K. H. Bouhadir, D. S. Hausman and D. J. Mooney, Polymer, 1999, 40, 3575–3584 CrossRef CAS.
- J. P. Draye, B. Delaey, A. Van de Voorde, A. Van Den Bulcke, B. Bogdanov and E. Schacht, Biomaterials, 1998, 19, 99–107 CrossRef PubMed.
- J. McCann, J. M. Behrendt, J. F. Yan, S. Halacheva and B. R. Saunders, J. Colloid Interface Sci., 2015, 449, 21–30 CrossRef CAS PubMed.
- S. K. Tripathi, R. Goyal and K. C. Gupta, Soft Matter, 2011, 7, 11360–11371 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional synthetic procedures, NMR, SEC, DLS, CLSM/DIC and electron microscopy data. See DOI: 10.1039/c7py01214d |
| This journal is © The Royal Society of Chemistry 2018 |