
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transiently malleable multi-healable hydrogel nanocomposites based on responsive boronic acid copolymers†
Adérito J. R.
Amaral
 ,
Mina
Emamzadeh
and
George
Pasparakis
*
,
Mina
Emamzadeh
and
George
Pasparakis
*
UCL School of Pharmacy, University College London, 29-39 Brunswick Square, London WC1N 1AX, UK. E-mail: g.pasparakis@ucl.ac.uk
First published on 10th January 2018
Abstract
The design of materials that mimic aspects of self-healing, as observed in nature, is of paramount importance as they could solve a number of problems in the biomedical field, such as the elimination of (bio-)material failure after prolonged use, improved integration at the interface with living tissues and more robust biomechanical performance. In this report, we propose the fabrication of soft hydrogels comprising a polymer matrix of poly(vinyl alcohol) mixed with a thermoresponsive boronic acid copolymer that is subsequently impregnated with poly(vinyl pyrrolidone) coated gold nanoparticles. The gels are crosslinked by the formation of reversible boronate ester bonds that can be remotely disrupted by a thermal or optical stimulus, endowing the gels with thermally-induced transient malleability that is optically trackable. It is demonstrated that the gels exhibit excellent healing properties within minutes even without the application of external stimuli. The rapid formation of the gels in concert with their fast and mild gel–sol phase transition under biologically relevant conditions is demonstrated by the encapsulation and release of cells in vitro. The proposed materials constitute a versatile cytocompatible platform for the construction of remotely healable soft gels for biomedical applications.
Introduction
Self-healing is an intrinsic property of living organisms that allows them to sustain their homeostasis in hostile environments in the event of injury.1 Evolution has driven the development of intricate healing mechanisms that combine a multitude of (bio-)chemical reactions and signals in an extremely synchronised manner that endows reliable recovery of key functions of damaged tissues to their initial functional state. From a materials point of view, it is possible to mimic certain aspects of self-healing towards the construction of healable materials that can repair microcracks automatically and autonomously.2–4In recent years, interesting chemical strategies have emerged to embed self-repair properties in bulk materials, which include the incorporation of healing agents in the form of nanoparticles/capsules or the introduction of dynamic/reversible chemical bonds as structural elements of the bulk material.5–8 The latter approach is particularly interesting and challenging in that suitable chemistries must be adopted that do not compromise the end-functionality of the designed material. Notable approaches of this method include the construction of healable polymer networks based on the exploitation of hydrogen bonds,9–11 π–π stacking type of interactions,12,13 host–guest recognition events,14–17 electrostatic interactions,18,19 metal coordination,20–22 acylhydrazones,23,24 reversible (hetero) Diels–Alder reaction motifs25–28 and disulfide bonds.29–31 Indeed, all these approaches have their own merits, however, the healing process is not always ex vi termini perfect as autonomous/automatic healing must be accompanied by gravitational force or mechanical/manual rejoining of the damaged material. In this regard, remotely controlled healing strategies allow for on-demand repairing of the material at the site of interest, although the need for mechanical intervention is not completely eliminated.
In principle, stimuli-responsive phase change materials fulfil this requirement of controlled self-healing by remote activation once their physicochemical properties are optimally integrated with the desired functionality.32,33 Examples of such systems include the use of “smart” polymers,34–36 the impregnation of polymer matrices with magnetic37,38 and gold nanoparticles39–41 (as photothermal antennae) to create hybrid hydrogel composites. In particular, gold nanoparticles (AuNPs) exhibit unique optical and physicochemical properties, which can be controlled by changing their size, shape, surface chemistry or aggregation, and are non-toxic at certain size ranges and concentrations.42,43 Although there is substantial body of literature on sol–gel type of polymers and other forms of materials (i.e. low molecular weight gelators),44 their use in the context of (remotely) healable systems has not been fully exploited despite the apparent advantages that they might convey: (1) their soft nature can be designed to match the mechanical properties of soft tissues, which renders them attractive candidates for (injectable) biomaterials design and/or cell therapeutic implants; (2) they allow for (transient) malleability during their gel–sol transition implying that their capacity to flow at confined spaces could, potentially, eliminate the need for mechanical intervention during the re-joining/repairing step, and (3) in principle, they can be doped with suitable fillers either to enrich them with additional functionalities of higher complexity or to reinforce their mechanical properties.8,45
In this work, we report a simple approach towards the construction of remotely healable and transiently malleable soft gels based on the dynamic, albeit covalent, boronate ester bond formation. Boronic acids are well-known to form boronate esters with cis-diols in a pH dependent manner: boronate ester formation is favoured at alkaline pH (the pKa of boronic acid is ≈8.8)46 that favours the formation of the boronate ester in the tetrahedral form and is hydrolysed at acidic conditions. Boronic acids in the form of polymers and hydrogels have been widely used in the biomedical field,47–50 for example, as pH-responsive hydrogels,51–54 carbohydrate biosensors,55–57 cell encapsulation systems,58 and as targeting motifs for drug delivery and diagnostics.59,60 Recently, soft healable gels based on the reversible bonding of boronic acids with diols have also emerged in the form of multi-responsive polymer blends.61–64 Inspired by these studies and our previous work on boronate ester constructs for chemical logic operations65 and cell surface engineering,66,67 we designed a simple boronic acid-based nanocomposite that exhibits complex, albeit predictable, physicochemical and mechanical behaviour as a multi-healable model platform for cell encapsulation. Our proposed system comprises a synthetic thermoresponsive boronic acid copolymer that is crosslinked with poly(vinyl alcohol) (PVA) to form cytocompatible hydrogels within seconds under physiological conditions (Fig. 1a). The network can further be impregnated with colloidally stable AuNPs that render the gels optically active without compromising their mechanical properties significantly. The gel ensembles can undergo reversible gel–sol transition owing to the disruption of the boronate ester crosslinks by thermal (heating above 37 °C), or optical (irradiation with green light) stimulus within seconds by taking advantage of the photothermal effect of AuNPs41,68 (Fig. 1b). The transient malleability of the material during the transition process can be conveniently monitored by an optical signal that is spatiotemporally confined at the stimulated areas, allowing for visual inspection of the recovered area. Interestingly, the reconstituted gels can fully recover their mechanical properties within minutes even without the application of external stimuli. Finally, we demonstrate that these materials can be used as bulk cell encapsulants for cell delivery applications aimed to soft tissue engineering applications,69–72 owing to the mild mixing and recovery conditions for their preparation and reconstruction. Such functionalities in terms of multi-responsiveness and healability, combined with excellent cytocompatible properties, have not been extensively exploited in previous boronic acid-based studies,33,60,64,73 and hence, we anticipate that our study will be inspirational towards the development of functionally more complex, yet synthetically accessible, materials for the biomedical field.
 | ||
| Fig. 1 Schematic illustration of (a) the formation of the hydrogels by their building components and (b) their (remotely triggered) self-healing properties. | ||
Experimental section
Materials
All the reagents and solvents were purchased from Sigma-Aldrich unless otherwise stated and used as supplied. Acetone (Fisher), acrylamide (AAm; ≥98%), 3-(acrylamido)phenylboronic acid (APBA; 98%), alizarin red S (ARS), 2,2′-azobis(2-methylpropionitrile) (AIBN; 98%), CellTracker™ Green CMFDA dye (Life Technologies), deuterium oxide (D2O; 99.9% D), diethyl ether (≥99.5%), dimethyl sulfoxide (DMSO; ≥99.9%), dimethyl sulfoxide-d6 (DMSO-d6; 99.9% D) (Cambridge Isotope Laboratories), Dulbecco's modified Eagle's medium (DMEM) (Gibco), phosphate buffered saline (PBS), N-isopropylacrylamide (NIPAAm; 97%), D-(+)-glucose (≥99.5%), ethanol (96%) (Fisher), foetal bovine serum (FBS), gold(III) chloride trihydrate (≥99.9% metal traces basis), penicillin–streptomycin solution, poly(vinyl alcohol) (PVA; Mw 146![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–186000 Da), poly(vinylpyrrolidone) (PVP; Mw 10000 Da), resazurin sodium salt, tetrahydrofuran (THF; ≥99.9%) (Fisher), and 0.25% (w/v) trypsin–ethylenediaminetetraacetic acid (EDTA) solution.
000–186000 Da), poly(vinylpyrrolidone) (PVP; Mw 10000 Da), resazurin sodium salt, tetrahydrofuran (THF; ≥99.9%) (Fisher), and 0.25% (w/v) trypsin–ethylenediaminetetraacetic acid (EDTA) solution.
Synthesis of poly(NIPAAm-co-APBA-co-AAm) (P1)
The copolymer was prepared according to a previously reported procedure.67 Briefly, NIPAAm (0.97 g, 8.6 mmol), AAm (92 mg, 1.3 mmol) and APBA (19 mg, 0.1 mmol) in a molar ratio of 86:13:1 (respectively) were dissolved in a 1:1 (v/v) DMSO/ethanol mixture (5 mL) in a 25 mL round-bottom flask. AIBN (16 mg, 0.1 mmol) was added to the flask, which was then sealed with a rubber septum before purging with argon for 10 minutes. The reaction mixture was placed in a pre-heated oil bath at 75 °C for 15 hours to initiate polymerisation under magnetic stirring. The polymer was recovered by exposing the reaction to room temperature, followed by precipitation in cold diethyl ether and drying under vacuum. P1 was isolated as a white powder (yield 79%, SEC Mn 17800 Da and Đ ≈ 2.1). The conversion rates of the NIPAAm and APBA monomers were monitored by 1H NMR by following the polymerisation reaction of a model poly(NIPAAm-APBA) copolymer with 80:20 NIPAAm:APBA initial monomer feed.
Synthesis of gold nanoparticles
PVP-capped AuNPs were synthesised based on a previously published procedure.74 In a typical reaction, a 0.02 M HAuCl4 aqueous solution (1 mL) was added to a freshly prepared aqueous solution of PVP (10 mL, 0.027 M) and left under vigorous magnetic stirring at 70 °C. The reaction was complete in 15–20 minutes as observed by the characteristic burgundy hue of the solution due to the formation of the AuNPs. The solution was cooled down to room temperature and the AuNPs were obtained by precipitation in acetone followed by washing in an ethanol solution. Finally, AuNPs stock solutions were dried and used throughout the experiments in the form of semi-solid pellets.Preparation of the hydrogels
Hydrogels were prepared from aqueous PVA (2.5, 5, 7.5 and 10 wt%) and P1 solutions (5, 7.5 and 10 wt%) in PBS (pH 7.4). The two components were mixed at equal volume ratios and mechanically stirred to ensure a homogeneous hydrogel formation at room temperature. The gelation time was monitored by a vial inversion test, which was completed in less than 10 s. AuNP-doped hydrogel composites were formed at different concentrations by dissolving the corresponding AuNPs pellet (20–40 w/v%) to the PVA solution, followed by mixing with the P1 solution.Mechanical characterisation and self-healing experiments
Rheological tests of the hydrogels were performed using a solvent trap to minimise sample drying. Oscillatory strain amplitude sweep measurements were performed at a frequency of 1 Hz to determine the linear viscoelastic region (LVR). Oscillatory frequency sweep measurements were then conducted at constant 10% strain amplitude to measure the storage (G′) and loss (G′′) moduli. The alternate step strain sweep of the hydrogels was measured at 25 °C and 1 Hz, switching from small strain (10%) to large strain (200%). The viscoelastic characteristics were also monitored by time and temperature sweep experiments at 10% fixed strain and frequency of 1 Hz, and the tanδ (G′′/G′) was determined.
Studies of the self-healing process and responsiveness of the hydrogels were also carried out by dynamic rheology. Hydrogel samples were cut in two pieces and gently put back in close contact for predetermined time intervals, at 25 °C, before measuring their recovered mechanical properties. A similar set of measurements was conducted on gels that were healed thermally (approximately 39 °C) or optically with green light, at predetermined time intervals, that allowed for full gel–sol–gel transition during the healing process. Then, G′ and G′′ vs. frequency and time of the reformed hydrogels were recorded. Self-healing efficiency was calculated as the ratio of G′ of the healed samples over the original values (time sweep at 25 °C and 1 Hz).
Cell culture
H9c2(2-1) (ATCC® CRL-1446™) cells (rat heart myoblasts) were maintained at 37 °C, under a 5% CO2 humidified atmosphere, in DMEM supplemented with 10% (v/v) heat-inactivated FBS, penicillin (100 units per mL) and streptomycin (100 μg mL−1). The cells were passaged every 2–3 days and reseeded prior to use.Cytotoxicity assays
Hydrogels were prepared as described above and sterilised by exposure them to ultraviolet (UV) light. The cytotoxicity of the constructs was assessed by an extraction test according to ISO 10993-5 standard as described by previously published studies.64,75,76 The gels were immersed in DMEM at an extraction ratio of 1 cm3 per 1.25 cm2 of gel surface area and incubated for 24 hours. Cells were then plated on a 48-well plate at a density of 4 × 104 cells per mL and incubated with the extraction fluid for 24 or 48 hours. The cell viability of the polymer and the hydrogels was assessed by the resazurin assay. Briefly, the cells were incubated with DMEM containing 2% (v/v) resazurin dye. After 2 hours, the absorbance (A) of the culture medium was measured at 570 and 600 nm. The cell viability was calculated as the percentage of untreated control cells, according to the formula: (A570 − A600) of treated cells × 100/(A570 − A600) of control cells.Cell encapsulation and release
H9c2 cells were stained with CellTracker™ green dye (according to the manufacturer's protocol), and suspended in the P1 solution (10 wt%). This cell suspension was then mixed with the PVA solutions (7.5 wt%), with or without AuNPs, until the gel formation was visually confirmed (under 10 s). Cell-laden hydrogels (1 × 105 cells per mL) were finally incubated for 1 or 2 days, and the culture medium was replaced every 12 hours. The cells’ morphology was observed under a phase-contrast microscope. Thereafter, the cell-loaded gels were dissociated (with glucose, temperature or light stimuli), and the recovered cells were resuspended in fresh medium and cultured for 2 days (without any polymer) to evaluate their viability through the resazurin assay.Statistical analysis
The data are presented as mean ± standard deviation (SD) and analysed using the SPSS Statistics software (version 22). The statistical significance of the differences was evaluated by one-way ANOVA using Bonferroni or Games-Howell post hoc tests. The level of significance was set at probabilities of *p < 0.05.Results and discussion
Synthesis and characterisation of poly(NIPAAm-co-APBA-co-AAm)
The copolymer was synthesised by free radical polymerization of N-isopropylacrylamide (NIPAAm), 3-(acrylamido)phenylboronic acid (APBA) and acrylamide (AAm) monomers. The final polymer was characterised by proton nuclear magnetic resonance (1H NMR) and size-exclusion chromatography (SEC); the Mn of P1 was found to be ca. 17800 Da. From the NMR spectrum, it was possible to determine the ratio of each monomer on the polymer backbone, which was found to be NIPAAm:APBA:AAm 87:2:11. The chemical composition of P1 was also verified by the characteristic aromatic peaks of APBA around 7.2–7.7 ppm and the isopropyl residue peaks of NIPAAm at 1.0 and 3.7 ppm in the NMR spectrum (Fig. S1a, ESI†). The lower critical solution temperature (LCST) onset of P1 was determined by turbidity studies using UV/Vis spectroscopy. The phase transition temperature onset was found to be 39 °C and 42 °C in phosphate-buffered saline (PBS) and Dulbecco's modified Eagle's medium (DMEM), respectively (Fig. S2a†). The rationale of the P1 design lies on preliminary experiments where it was found that the incorporation of APBA, shifted the LCST onset to lower values due to its relative hydrophobic character; hence, the copolymerisation with the hydrophilic AAm monomer, compensated for the LCST reduction and resulted in a shifting of the LCST onset within a therapeutically/clinically relevant temperature window77,78 (i.e. slightly above the physiological temperature) to potentially facilitate in/ex vivo biomaterial intervention. In addition, the sharp shifting of the LCST onset by the incorporation of the two co-monomers, implies the relatively random distribution of the co-monomers within the polymer backbone, reflecting homogenous conversion rates for all monomers;79,80 this was demonstrated by monitoring the diminution of the 1H NMR signal of the vinyl protons of APBA (5.7, 6.3 and 6.45 ppm) and NIPAAm (5.5, 6.08 and 6.2 ppm) during the polymerisation of a model poly(NIPAAm-co-APBA) copolymer, where it was confirmed that NIPAAm was only slightly more reactive than the APBA residues (10% more reactive in the first hour of the reaction, Fig. S1b†). Finally, the cis-diol capturing capacity of the APBA residues (even at pH 7.4, which is below the optimum pKa for boronate ester formation) was conveniently shown in turbidity experiments conducted in aqueous solutions (Fig. S2b†). The glucose-boronate esters formed increase the overall solubility of the polymer, which shifts the LCST onset to higher temperature,81,82 as observed in DMEM.
Preparation and physical characterisation of the gold nanoparticles
Poly(vinyl pyrrolidone) (PVP) stabilised AuNPs were prepared via a one-step process, and characterised by UV/Vis spectroscopy, dynamic light scattering (DLS) and zeta potential. The UV/Vis spectrum of the PVP coated AuNPs showed a characteristic surface plasmon resonance (SPR) band at ca. 520 nm (Fig. S3a†). The TEM images revealed relatively homogenous and spherical particles with an average diameter of 4.5 ± 1.3 nm (Fig. S3c†). As expected, the DLS studies showed a significantly higher z-average diameter of 15.14 ± 0.35 nm (with PDI of 0.52 ± 0.02), which is expected due to the hydrodynamic shell of the PVP layer in solution. The AuNPs exhibited an almost negligibly negative zeta potential (−3.18 ± 0.23 mV), due to the oxidized sites of the PVP that occur during the reduction of the gold salts.74,83 PVP was chosen as a steric stabiliser of the AuNPs as it is biocompatible and provides excellent colloidal stability within a wide range of pH values and ionic strength.42 Moreover, it allows for excellent mixing of the AuNPs with the other components of the gels without noticeable aggregation during the hydrogel preparation.Formation of the hydrogels
The hydrogels were prepared simply by mixing aqueous solutions of P1 and PVA (PBS, pH 7.4) at 25 °C; the gel was formed almost instantly, within 10 s after the mixing of the two precursor polymeric solutions, owing to the spontaneous formation of covalent but reversible boronate ester bonds between the boronic acid moieties of P1 and the diol residues of PVA.57,84 It should be noted that although the optimum pH for the formation of boronate ester complexes is close to 9 (pKa ≈ 8.8) at physiological conditions, there is still sufficient amount of ionisable boronic acid groups (ca. 6% of the total boronate moieties) that bind to cis-diols present on the PVA component at pH 7.4.81,85 This is also demonstrated by the fast gelation times as well as the rapid healing mechanism, which is further augmented by the abundance of cis-diols residues provided by PVA. Preliminary experiments were conducted by mixing different concentrations of P1 and PVA to determine the minimum polymer feed required to form self-standing gels (Table S1†). Optically active hydrogels were prepared by adding PVP-coated AuNPs in the PVA solution before mixing with the P1 precursor solution. These gels had a characteristic red/burgundy colour, indicative of the homogeneous distribution of the AuNPs within the polymeric network. Hydrogels of different optical densities could be formed by adjusting the AuNPs feed in the mixing step.The hydrogels – 5% P1–3.7% PVA (P1–PVA) and 5% P1–3.7% PVA–10% AuNPs (P1–PVA–AuNP) – were characterised by UV/Vis spectroscopy, Fourier transform infrared (FT-IR) spectroscopy, thermogravimetric analysis (TGA) and scanning electron microscopy (SEM). The UV/Vis spectrum of the AuNPs doped hydrogels showed a characteristic SPR band at ca. 520 nm with slight broadening (Fig. S4†), possibly due to partial aggregation of the AuNPs during the hydrogel preparation.86 As expected, the non-doped hydrogels showed no apparent absorbance over the wavelength range from 400 to 700 nm.
Fig. 2a shows the FT-IR spectra of the freeze-dried samples of the building components of the gels, namely, P1, PVA, PVP, P1–PVA and P1–PVA–AuNP. Characteristic peaks that appear in P1 and PVA also appear in both gels: a strong amide I band at 1630 cm−1 and amide II at 1530 cm−1 of the P1 poly(acrylamide) backbone; a broad band around 3300 cm−1 corresponding to the free –OH groups of PVA and free water residues. Typical peaks of the isopropyl group of PNIPAAm at 1360 and 1320 cm−1 appear in both P1–PVA and P1–PVA–AuNP gels; in addition, the bands at 1430 and 1010 cm−1 can be associated with the stretching vibration of the phenyl ring, while the peak at ca. 1290 cm−1 can be assigned to the B–O stretching.87,88 The distinctive absorption peaks (broad) at 3100–3500 cm−1 and 2800–2950 cm−1 correspond to –NH, and –CH3/–CH groups of P1, respectively. The characteristic C–N stretching of PVP at 1260 cm−1 is also present in the P1–PVA–AuNP spectrum.
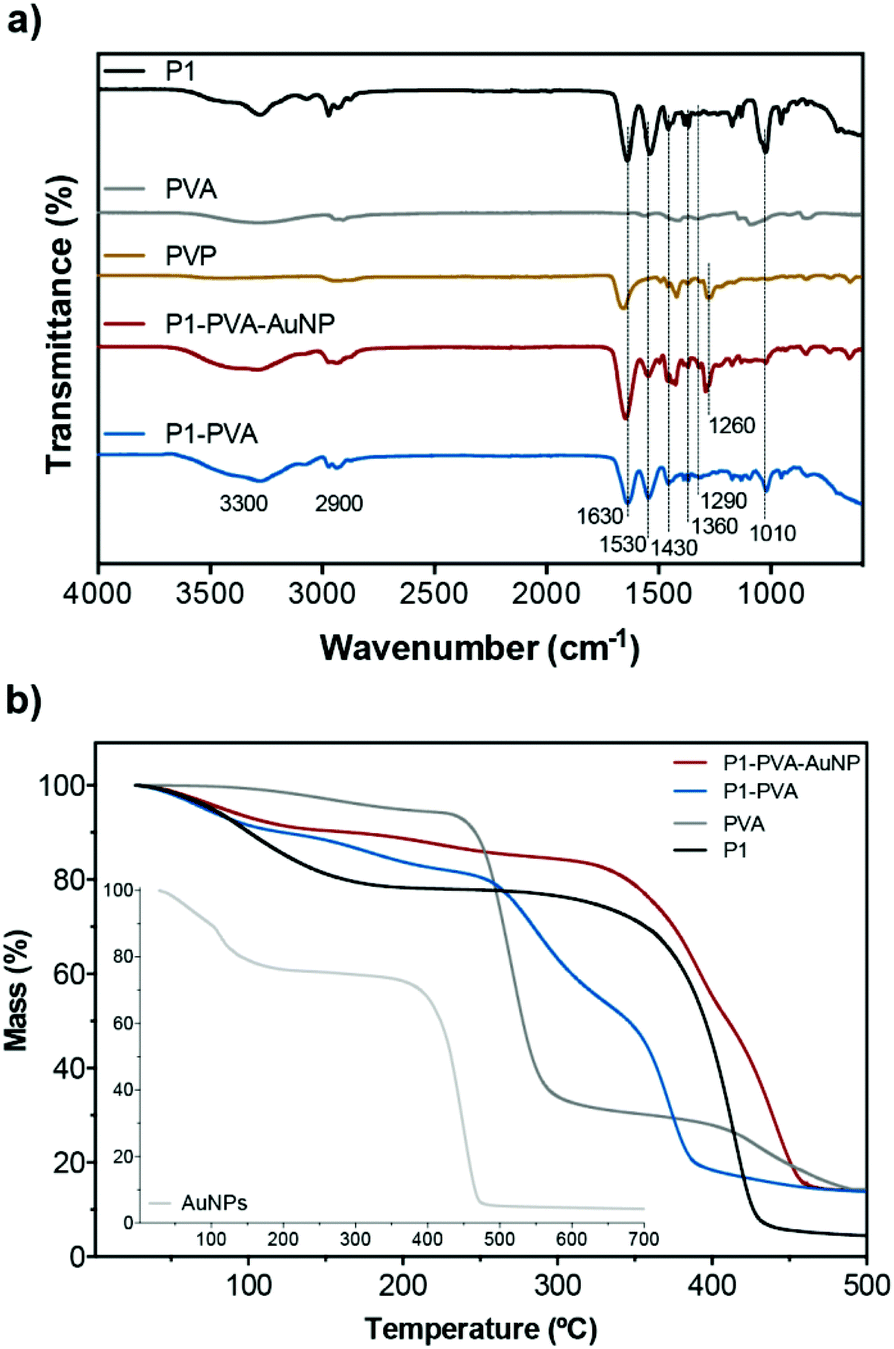 | ||
| Fig. 2 (a) FT-IR spectra and (b) TGA profiles of the hydrogels and their components. | ||
The TGA traces (Fig. 2b) display that P1 exhibits a two-phase mass loss, one from 30–200 °C due to moisture and other volatiles, and a main thermal degradation profile with onset around 270 °C, which is completed at 440 °C (typical of PNIPAAm). PVA exhibits minor mass loss from 100 to 260 °C, followed by a sharp mass loss up to 340 °C, and a third phase of lower mass loss rate that is completed at 500 °C. As expected, the P1–PVA gel exhibits a degradation profile that follows the average degradation profiles of P1 and PVA, which is indicative of good polymer blending during the hydrogel preparation process. The P1–PVA–AuNP gel exhibits a significant thermal degradation resistance owing to the thermally stable PVP-coated AuNPs that show thermal degradation onset at 370 °C. Fig. S5† depicts a typical SEM image of a freeze-dried hydrogel, where it can be seen that a characteristic fibrous-type, micron-sized porous network has been formed.
The hydrogels showed interesting thermal responsiveness owing to the PNIPAAm content of P1. To fully understand the thermally induced gel–sol transition we conducted preliminary 1H NMR studies of dilute mixtures of P1 with PVA at different temperatures (Fig. S6a†). When the temperature was raised to 40 °C, the intensity signal of the CH(CH3)2 (at 3.6 ppm) and –CH(CH3)2 (at 1.1 ppm) peaks of PNIPAAm was significantly reduced as a result of the coil-to-globule transition of P1 above the LCST. More importantly, the peaks related to the aromatic group of APBA at ca. 7.2–8.0 ppm and the backbone (at 2.0 ppm) were also attenuated, while the signal intensity of characteristic PVA peaks at 3.8 and 1.5–1.8 ppm remained unaffected. It is therefore reasonably concluded that the boronate ester formation should be more favourable at temperatures below the polymer's LCST where the APBA residues are more solvated, while a disruption of the boronate ester bond should be favoured when P1 exists in a globule/collapsed state. A more detailed insight of the actual thermally induced binding and release of the PVA diols from P1 was studied by employing a fluorescence assay based on the diol-containing dye alizarin red S (ARS).89–91 ARS is inherently non-fluorescent but fluoresces when bound to boronic acid residues.92 Mixing of P1 with ARS in alkaline glycine buffer at room temperature resulted in the formation of a characteristic orange solution indicative of the formation of the fluorescent P1–ARS complex (Fig. S6b†). The complex formation could be disrupted simply by raising the temperature at 45 °C (above the LCST), resulting in a characteristic change of colour of the solution to pink (with a slightly turbid hue due to the collapsing of P1), which is the non-boronate bound colour of the initial non-fluorescent ARS, suggesting the release of the ARS molecules from P1. It was possible to monitor the process of the disruption of the P1–PVA complexes by measuring the variation of the fluorescence intensity of the boronate–ARS complex as a function of temperature (Fig. S6b†) and time (Fig. S6c†); a gradual decrease of the fluorescence intensity was observed by increasing the temperature (and vice versa), which became sharper above ca. 38–40 °C. In addition, by keeping the temperature constant at 45 °C, it was found that the ARS was released from P1 in less than 4 minutes, as evidenced by the sharp decrease of the fluorescence intensity, which is in good agreement with the required timeframe to induce macroscopic gel–sol transition of the synthesised gels (see below). Previous works demonstrated that ARS molecules were chemically released from similar thermosensitive PBA-containing copolymers89,90 and hence, this set of experiments enabled us to understand the proposed mechanism of the temperature controlled diol release from P1 and to harness it in order to construct P1–PVA gels.
By doping the gels with AuNPs, it allowed us to trigger this thermally activated gel–sol type of transition via an optical switch by irradiating the gels at the SPR band (i.e. with monochromatic green light at 521 nm). We examined the relationship between the concentration of AuNPs in the matrix and the time required to induce complete gel–sol transition (Fig. 3a and b). As expected, the phase transition time of the nanocomposites reduced proportionally with the increase concentration of AuNPs incorporated in the matrix due to the enhanced light-to-heat conversion rates induced by the AuNPs.93 More importantly, it was possible to induce full gel collapsing at around 39 °C within 40 s (AuNP10), which could be further enhanced to ca. 25 s for the AuNP20. As expected, no phase transition was observed in control P1–PVA gels after light irradiation (Fig. S7†). The thermally or optically disrupted gels can be seen to flow under gravitational force in Fig. 3c (above the LCST by direct heating or by light irradiation). Upon removal of the thermal or the optical stimuli, the gels recover very rapidly, within seconds, to their initial state. This rapid transition even in bulk allowed us to form gel samples that could be cut, and then re-joined by bringing them in direct contact. Although the diol-boronate ester type of crosslinking constitutes a facile covalent, albeit dynamic, type of bonding and allows the re-constitution of cut/damaged samples, the application of the thermal stimulus either directly (i.e. for P1–PVA) or by light irradiation (i.e. for P1–PVA–AuNP gels), allows for a faster recovery/healing process as well as for nearly complete restoration of key mechanical properties in very short time-frames. Moreover, the transient gel–sol transition phase during the application of the thermal or the optical stimulus renders the gels malleable, allowing for tailor-made re-shaping in a highly controllable manner.
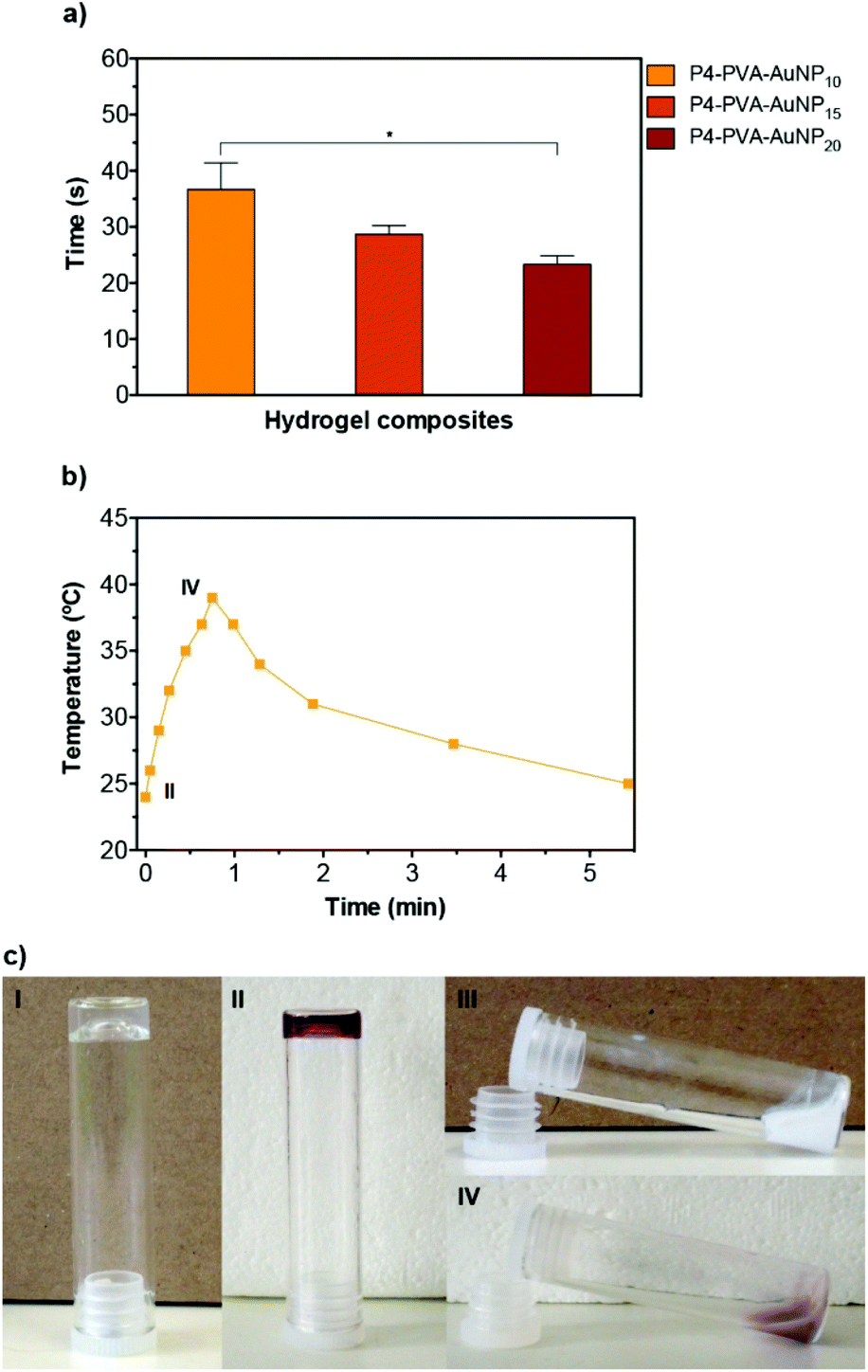 | ||
| Fig. 3 (a and b) Kinetics of P1–PVA–AuNP gels’ phase transition with different concentrations of AuNPs (10–20%) after light irradiation at 521 nm. The data are expressed as mean ± SD from three experiments (*p < 0.05). (c) Vial inversion tests of P1–PVA and P1–PVA–AuNP hydrogels demonstrate temperature (I, III) and light (II, IV) dependent viscoelastic mechanical properties. | ||
Mechanical properties of the polymer gels
Further, we studied the dynamic rheological properties of the hydrogels at representative P1, PVA and AuNPs concentrations. Initially, oscillatory strain sweeps were carried out at 25 °C to determine the linear viscoelastic region (LVR) of the hydrogels. The storage (G′) and loss (G′′) moduli were found to intersect at a critical strain value of 110% and 80% for P1–PVA and P1–PVA–AuNP, respectively, which is required to disrupt the polymeric network of the gel and induce a transition to a fluidic state beyond this point (Fig. S8†). These results suggest that the free-standing hydrogels could withstand relatively large deformations, which was also evidenced by their ability to stretch into long thread-like dimensions, similar to a “spinnbarkeit behaviour”94 (Fig. S9†).Then, frequency sweep measurements were performed within the LVR to evaluate the mechanical strength of P1–PVA and P1–PVA–AuNP gels, as seen in Fig. 4a. At high frequency regimes, the G′ was higher than the G′′ for both gels, indicating a gel-like character, and the elastic component dominates. The crossover frequency point between G′′ and G′ for P1–PVA and P1–PVA–AuNP was ca. 0.48 Hz and 0.65 Hz, respectively, below which the gels show a liquid-like behaviour as the viscous component dominates, allowing time for the network to restructure itself under stress.14,94 As expected, these values suggest that the impregnation of the gels with AuNPs somewhat compromises their mechanical properties, which can be attributed to the fact that doping results in a lower crosslinking density per gel weight.
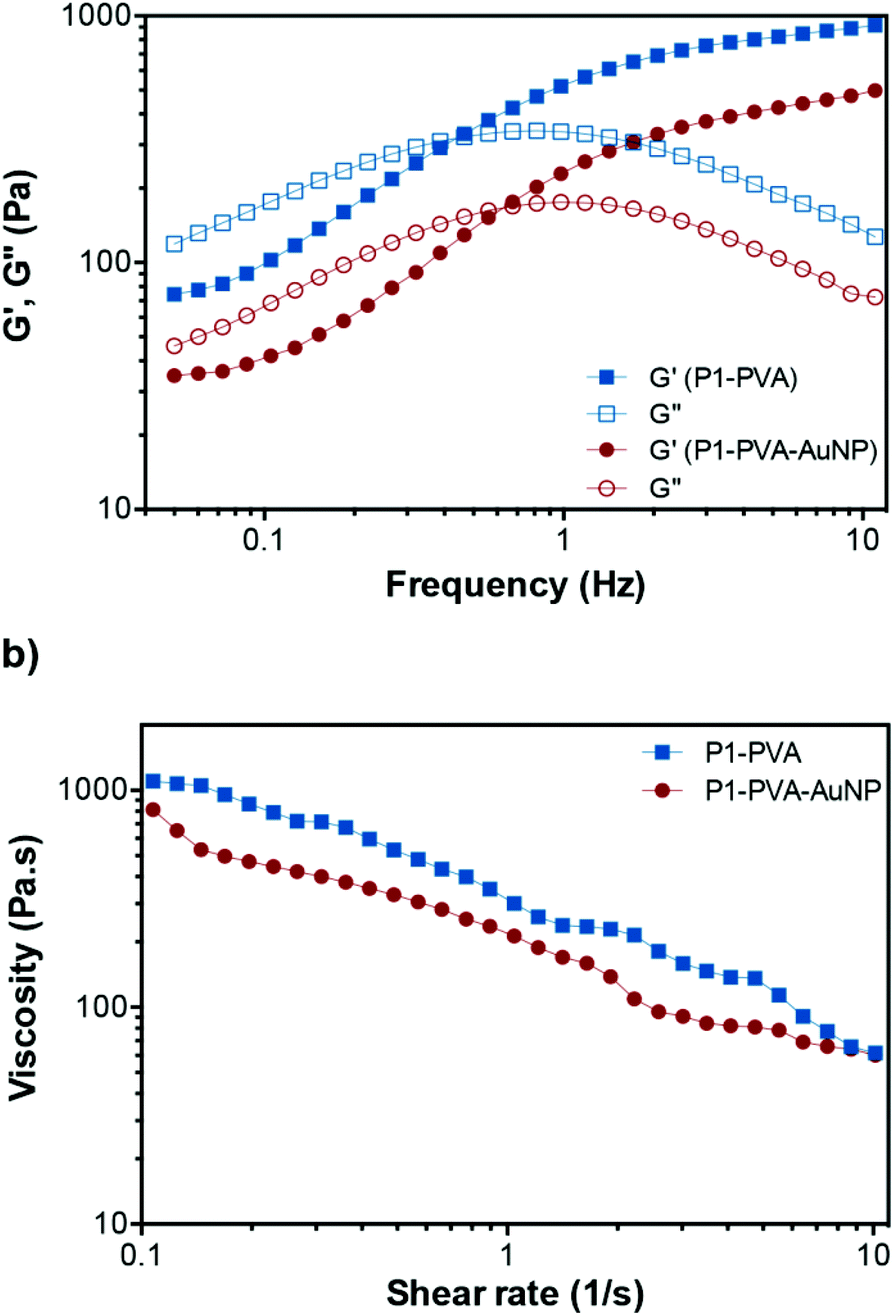 | ||
| Fig. 4 Rheological properties of the hydrogels. (a) Variation of the G′ and the G′′ of P1–PVA and P1–PVA–AuNP as a function of frequency. (b) Shear-thinning behaviour of the hydrogels at 25 °C (10% strain). | ||
Next, we studied the shear-thinning properties of these gels. The viscosity of both P1–PVA and P1–PVA–AuNP at 25 °C decreased almost linearly with the shear rate increasing as the dynamic boronate ester crosslinks in the network start to disrupt (Fig. 4b). As previously mentioned, these hydrogels demonstrated a frequency-dependent viscoelastic behaviour, which is characteristic of dynamic gel networks with reversible covalent bonds.76,94,95 To clarify the influence of different polymer concentrations on the rheological properties of the hydrogels, we compared the G′ and G′′ of the gels with different P1/PVA/AuNPs stoichiometry. It was found that when the concentration of both P1 and PVA decreased from 5 wt% to 2.5 wt%, the G′ decreased dramatically from 800 to 90 Pa, apparently due to the lower crosslinking density that results in faster relaxation of the polymer network after oscillatory perturbation (Fig. S10a†). The same trend was observed when we kept the concentration of P1 constant (at 2.5%, 3.7% or 5%), but reduced the PVA feed from 5% to 3.7% and 2.5%: a linear decrease of the G′ and the G′′ was observed in all cases, indicative of the impact of crosslinking density on the mechanical properties of the gels (without changing their reversible nature). A linear decay of the G′ and the G′′ was also observed by increasing the AuNPs feed from 10% to 20% (Fig. S10b†). Therefore, we shortlisted the P1–PVA–AuNP10 as the optimum sample for further testing owing to its excellent optical properties – that is a rapid photo-triggered gel–sol transition within ca. 40 seconds – and good mechanical stability.
The gels were found to exhibit temperature-dependent viscoelastic properties due to the thermoresponsive PNIPAAm element of P1. Temperature sweep tests were then performed on P1–PVA to study this effect (Fig. 5a). An increase of temperature from 25 °C to 40 °C resulted in a gradual decrease of the G′ and crossover with G′′ at approximately 39 °C (tanδ = 1), the critical gelation temperature, indicating a gel–sol transition.22 In a complete heating–cooling cycle (25–40–25 °C), Fig. S10c,†G′ and G′′ restored their original values, confirming the thermo-reversibility of the mechanical properties of the hydrogel, which is dependent on the thermal sensitivity of the equilibrium constant, and relates to the initial degree of crosslinking and bond enthalpy.94,96 When the hydrogel is formed, boronate ester crosslinks decrease the mobility of the polymer chains and hence the entropy. Below the critical temperature, the system is in the gel state because the binding enthalpy of the complex compensates for the loss of entropy. Further temperature increase, above this point, translates in the increase of the Gibbs free energy (ΔG = ΔH − TΔS) due to larger positive value of TΔS. The binding enthalpy cannot compensate this increment that results in the disruption of the gel.97,98 This is facilitated by the “hide-and-reveal” type of interaction that is driven by the well-known reversible coil-to-globule transition of NIPAAm copolymers in aqueous solutions.65,89,99
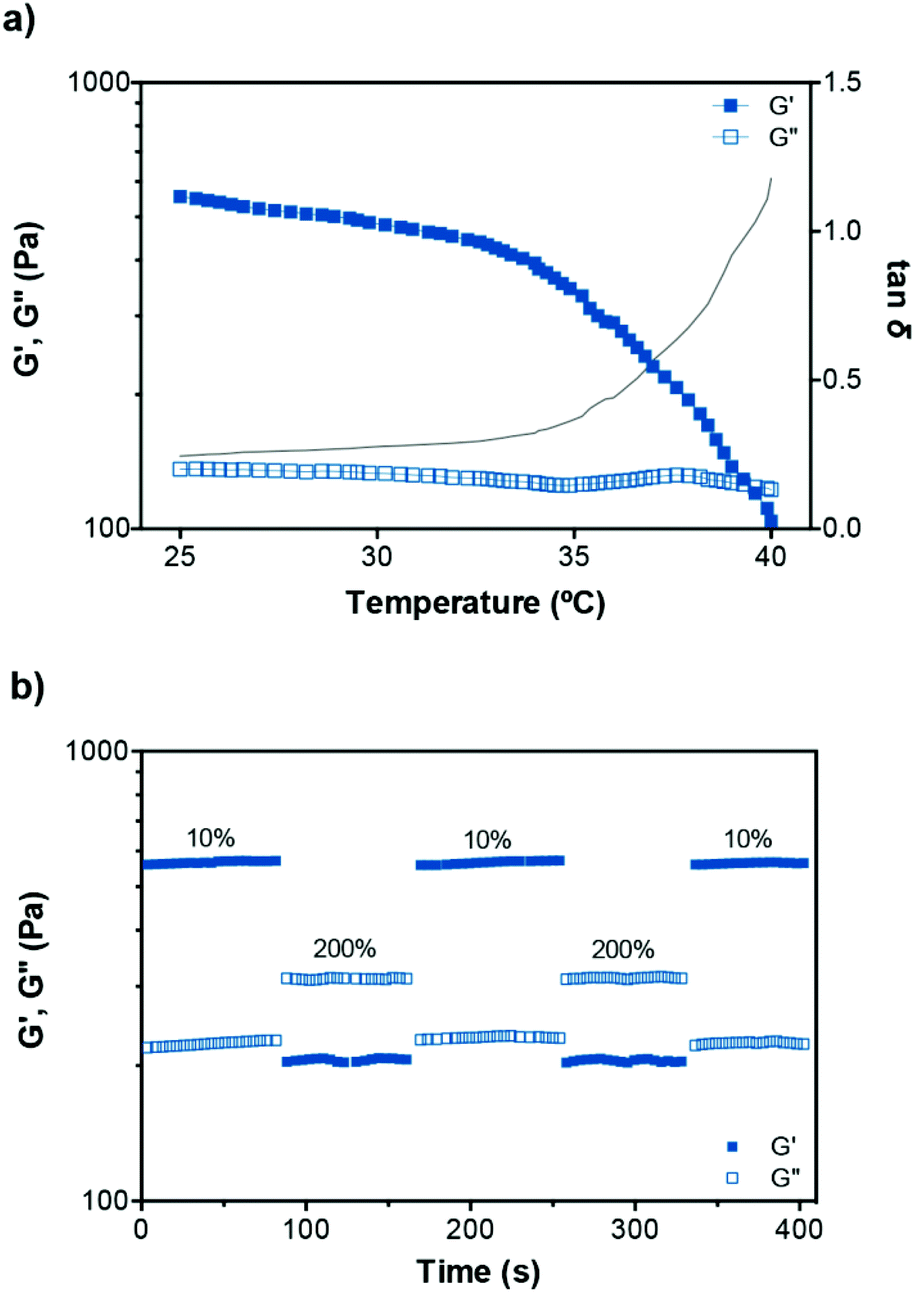 | ||
| Fig. 5 (a) Variation of the dynamic moduli and tanδ of P1–PVA with temperature. (b) Representative step-strain measurements of P1–PVA gel (25 °C, 1 Hz). | ||
To clarify the diol-boronate binding specificity, a PNIPAAm homopolymer was synthesised and mixed with PVA; as expected, no gel formation was observed. Another control experiment involved the immersion of a P1–PVA gel in a 2.5% (w/v) concentrated glucose solution where rapid degradation was observed (Fig. S11†). It is concluded that the gels do exhibit tolerance to low glucose content primarily due to the higher binding constant of P1 towards PVA compared to free glucose residues and to lesser extent to the slow diffusion of dilute glucose gradients within the gel's polymer matrix. These results highlight the glucose-sensitive nature of the hydrogels and the diol-boronate specific type of crosslinking.
Finally, we calculated the approximate mesh size (ξ) values of the hydrogels by using the experimental values of the G′ to calculate the approximate molecular weight between crosslinks (see ESI†).100,101 The ξ values for P15–PVA2.5 and P15–PVA3.7 hydrogels were found to be 5.07 ± 0.13 nm and 4.46 ± 0.09 nm, respectively. The mesh size gives a rough estimate of the global microstructure of the gel (although network defects, such as banging chain ends and slipping chain entanglements can be present), and can provide a relatively generic guide on correlating the mechanical properties with the crosslink density of the polymer network.102,103 Indeed, our reported ξ values are consistent with the measurements of reported gels of similar structure, texture, and mechanical properties,101,102,104 and further support the concept that the molecular microstructure directly impacts the macroscopic properties of the gels as evidenced experimentally.
Self-healing properties
To investigate the self-healing ability of the hydrogels, step strain measurements were conducted to determine the autonomous recovery of their mechanical properties following network rupture at high strains (see Fig. 5b). When the P1–PVA gel was subjected to low strain values (10%), the G′ became larger than the G′′. However, when a high magnitude strain was applied (200%), the G′ value decreased from ≈550 Pa to 220 Pa accompanied by a characteristic inversion of the G′ and the G′′, due to the disruption of the gel network. When the strain returned to 10%, both the G′ and G′′ of the gel recovered their initial values within seconds as a result of the rapid reconstitution of the dynamic crosslinks. The process could be repeated several times without any noticeable loss of the mechanical properties.Frequency sweep experiments of the healed gels with (temperature or light, Fig. 6a) or without stimulus (by mechanical rejoining at 25 °C, Fig. 6b) showed a comparable profile to the original materials. The healed samples exhibited negligible hysteresis of both the G′ and the G′′ throughout the frequency range tested, and similar crossover frequencies of the G′ and G′′ values. The largest hysteresis was observed in the AuNPs-doped samples that were healed at 25 °C without stimulus; however, their profile was significantly improved upon application of the optical stimulus during healing. We hypothesise that the PVP coating of the AuNPs may be inhibiting the kinetics of the healing process at room temperature although the recovery of the sample is almost fully restored by application of the optical stimulus, presumably due to the enhancement of the mobility of the polymer chains, which in turn improve the healing process kinetics. Overall, these results corroborate our hypothesis of the complete recovery of the mechanical properties of the gels owing to the covalent, albeit reversible nature of the boronate ester groups at the re-joining interface even in the absence of any stimulus.
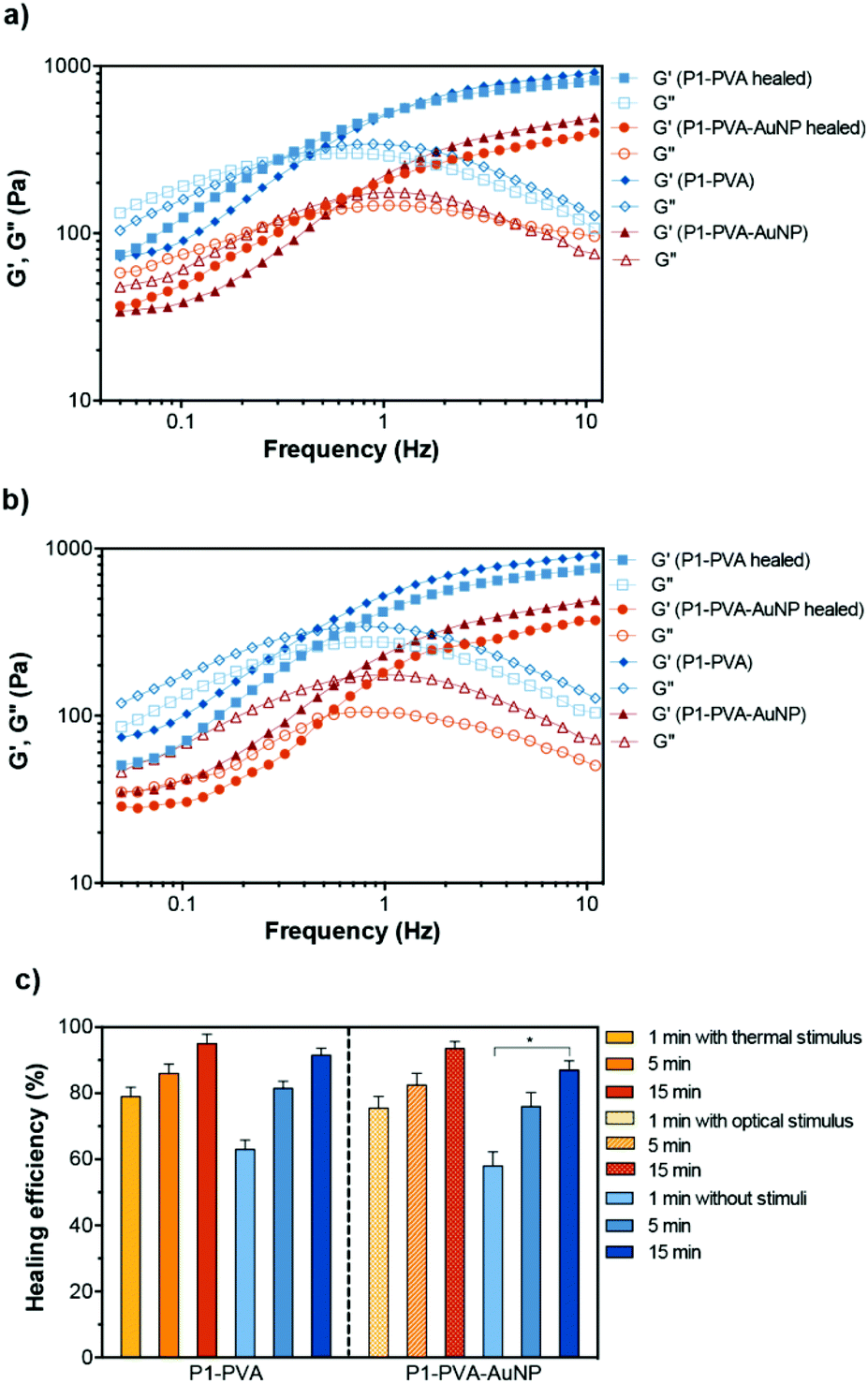 | ||
| Fig. 6 Frequency sweep of P1–PVA and P1–PVA–AuNP gels before and after (a) the stimuli-induced healing process (thermal and optical), and (b) after gel cut and rejoining (without stimulus at 25 °C). (c) The self-healing efficiency of P1–PVA and P1–PVA–AuNP in different conditions over time (mean ± SD from triplicates, *p < 0.05). | ||
The self-healing efficiency of P1–PVA and P1–PVA–AuNP was quantitatively evaluated by comparing the recovered G′ value of the samples after 1, 5 and 15 minutes with or without the application of a thermal (ca. 39 °C) or optical stimulus (by reforming the sample gel from initially cut and rejoined bulk pieces). The application of the thermal stimulus on the P1–PVA gel results in a more rapid recovery rate of the mechanical properties one minute after thermal treatment (79%) compared to the non-thermally treated sample (61%), presumably due to the augmented polymer chain mobility driven by the gel–sol network transition within this timeframe. Interestingly, both thermally and non-thermally treated samples (P1–PVA) recovered to almost 95% of their initial strength in 15 minutes (Fig. 6c). The same trend was observed for the optically treated sample, P1–PVA–AuNP: the sample recovered ca. 75% of its initial G′ value, compared to 60% of the non-optically treated sample, one minute after application of the optical stimulus. Again, the optically treated sample recovered more than 90% of its initial G′ within 15 minutes post-healing, although the non-optically healed sample recovered ca. 85%. It should be noted that regardless of the application of any stimulus, all the hydrogels exhibit remarkably rapid healing capabilities compared to other structurally and mechanically similar gels that require hours or even days for full recovery.24,62,105
Photographs of the thermal- and optical-initiated healing process, in Fig. 7, display the self-healing properties of the gels with or without the application of stimuli. The thermally rejoined gel (P1–PVA, Fig. 7a) exhibited a characteristic white hue in its sol state, due the coil-to-globule transition of P1, which in turn renders the gel malleable. This results in the spontaneous formation of a meniscus at the interface of the two cut samples that can be combined with minimum mechanical intervention to reform the healed sample (Movies S1 and S2†). The same behaviour was observed in the optically rejoined sample (P1–PVA–AuNP, Fig. 7b and Movie S3†) that exhibited a distinct purple hue due to the transient aggregation of the AuNPs due to the network collapsing. Again, the healed sample was spontaneously formed with no or minimum mechanical intervention. The samples that were repaired without the application of any stimulus (Fig. 7c and d and Fig. S12†) could also be fully healed, although the aesthetic result was less satisfactory as the cut interface was still vaguely visible just after mechanical rejoining (Movie S4†), presumably due to the reduced malleability of these samples. It is interesting to note that the transient colour change could be used as an optical signal to indicate when the gels are “malleable-ready” to perform reforming/healing procedures only when/where the stimuli are applied. In addition, the remote reforming of the gels at confined areas without mechanical/manual rejoining due to the transient gel–sol transition, allows for these materials to find applications as healable biomedical sealants, cavity fillers or soft scaffolds for tissue engineering.106,107
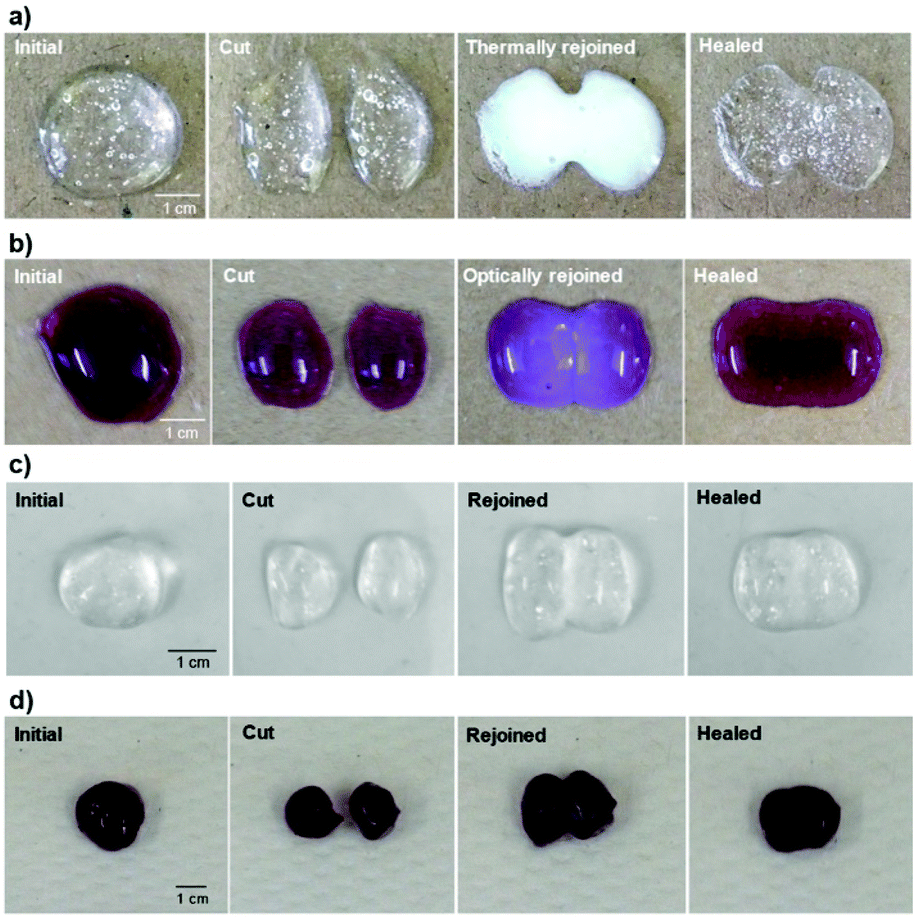 | ||
| Fig. 7 Thermally and optically triggered healing process of (a) P1–PVA and (b) P1–PVA–AuNP, respectively. Digital photographs of initial hydrogel samples of (c) P1–PVA and (d) P1–PVA–AuNP after cut through the centre and rejoined for self-healing into one integral piece at 25 °C without any external stimulus. The healed gel could maintain its integrity and withstand shaking. | ||
Cytocompatibility, cell encapsulation and release from the gels
Once the physicochemical and mechanical properties of the hydrogels had been characterised, we examined their biocompatibility and the possibility to use them as cell carriers. Cytotoxicity studies were performed to probe the effect of P1, P1–PVA and P1–PVA–AuNP on the viability of H9c2 cells using the resazurin assay. The copolymer P1 was found to exhibit excellent cytocompatibility for concentrations ranging from 25 to 1000 μg mL−1 (Fig. S13†). In addition, cells were cultured in the gel extracts for 48 hours and no cytotoxicity was found for all the gel batches tested with a cell viability above 80% (Fig. 8a).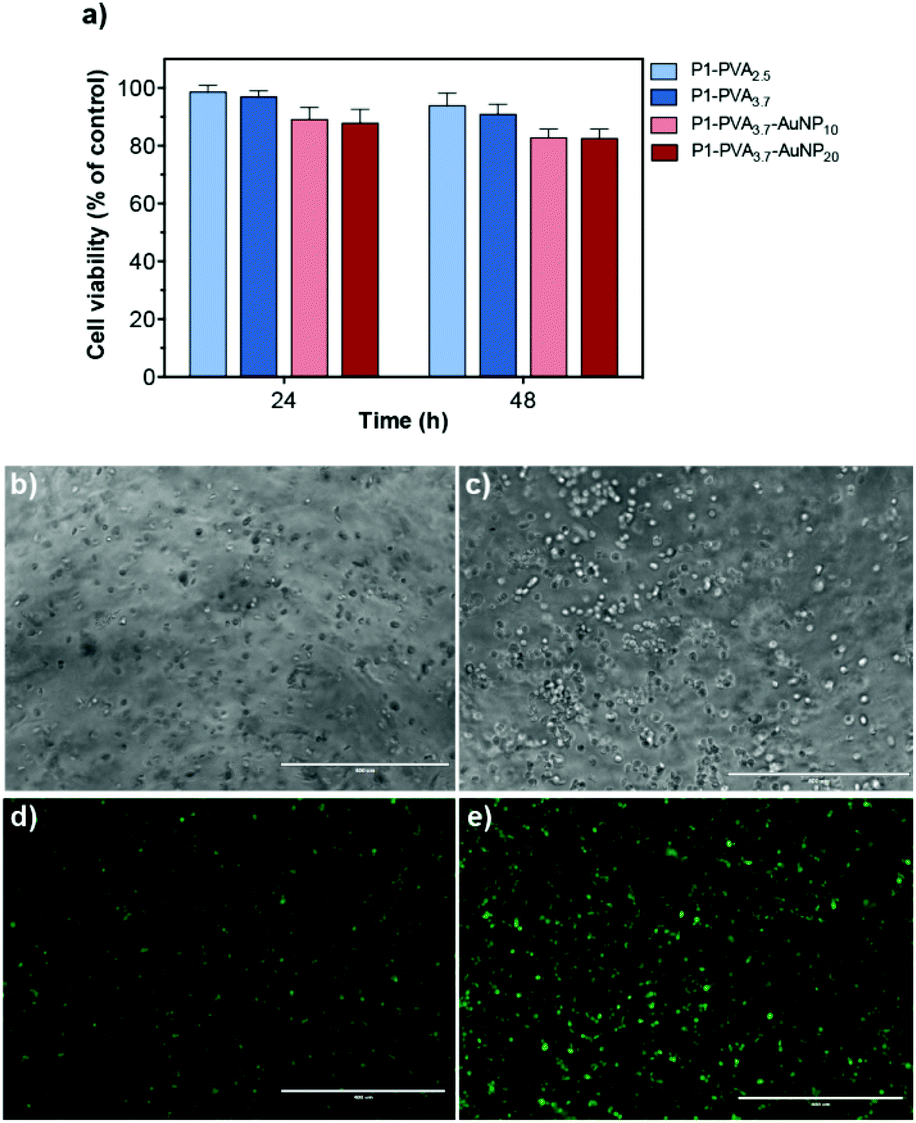 | ||
| Fig. 8 (a) Cell viability studies of P1–PVA2.5–3.7 and P1–PVA3.7–AuNP10–20. The cells were incubated in the extraction media containing the gels and the cell viability was measured using a resazurin assay at 24 and 48 h. The data are shown as a percentage of cell viability with respect to the control corresponding to untreated cells (mean ± SD obtained from triplicates). Microscopy images of H9c2 cells encapsulated in the P1–PVA hydrogel at (b) 24 h and (c) 48 h, and stained with CellTracker™ Green dye after (d) 24 h and (e) 48 h (z-axis projection; scale bars = 400 μm). | ||
Next, we sought to study the gels as cell encapsulants, as several studies have shown that soft hydrogels can support the retention of viable cells within polymeric matrices for cell delivery/therapy and tissue engineering applications.60,69,72,102,108–110 Optical microscopy images showed that H9c2 cells could spread evenly within the gel matrix, retained their morphology and apparently increased their density when cultured for 48 hours (Fig. 8b and c); also, the cells predominantly remained alive throughout the culture period as evidenced by the intense green fluorescent signal (Fig. 8d and e).
Subsequently, P1–PVA hydrogel was conveniently degraded by the addition of glucose solution (0.14 M) to disrupt the boronate ester crosslinks, or simply by slightly increasing the temperature (≈ 39 °C) to induce polymer network collapsing. Similarly, the P1–PVA–AuNP gel could be dissociated by short-time irradiation with green light, and demonstrated very good cytocompatibility (Fig. 9a). The released cells could be harvested and cultured on a new tissue culture well plate, where they could proliferate with time comparably to non-encapsulated cells (control group), without any deleterious effect on their morphology (Fig. 9b and c). It has been reported that hydrogels containing dynamic reversible crosslinks may promote the exchange of nutrients and metabolites between the cells and the external environment, and allow the cells to engage in complex cellular functions, which can represent an advantage in 3D cell culture compared with permanently crosslinked networks.95,111,112 Therefore we believe that our proposed materials could constitute a robust remotely healable platform to encapsulate and release cells with applications in cell delivery technologies.
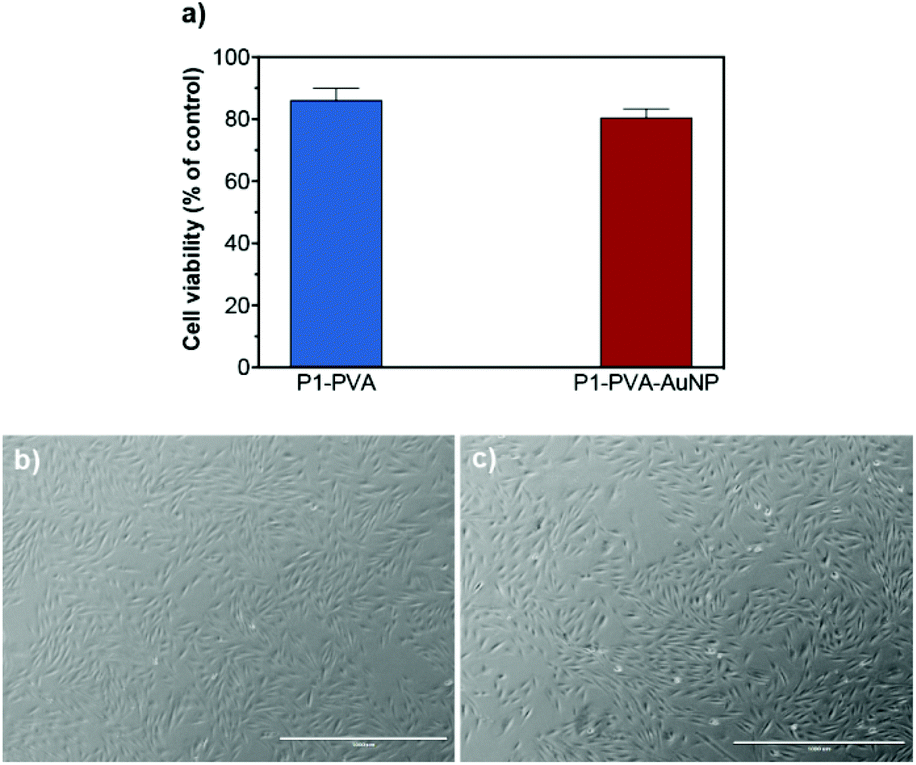 | ||
| Fig. 9 (a) In vitro cytotoxicity of H9c2 cells after being encapsulated and released (48 hours). Data presented as percentage of cell viability with respect to the control corresponding to non-encapsulated cells (mean ± SD obtained from triplicates). The cells after (b) P1–PVA and (c) P1–PVA–AuNP gel dissociation could proliferate for 2 days in culture at 37 °C (scale bars = 1000 μm). | ||
Conclusions
In summary, we have successfully designed multi-responsive self-healing hydrogels based on the dynamic covalent boronate ester complex formed between a thermoresponsive boronic acid-based copolymer and PVA. These constructs can further be doped with colloidally stable PVP-coated AuNPs to form optically active gel nanocomposites. The proposed materials comprise a photothermally induced gel–sol type of transition that can be remotely activated, providing transient malleability that can be spatiotemporally controlled by visual inspection of the stimulated areas. In addition, the mechanical and optical properties of the gels could be broadly tuned simply by adjusting the relative ratios of their building components. The photothermally controlled dynamic/reversible character of the boronate ester crosslinks as well as their intrinsic healing nature endows the gels with shear-thinning and rapid healing properties under biologically relevant conditions. Indeed, the proposed gels were capable to encapsulate cells, retain their viability for days and release them without apparent adverse effects on their viability. The versatile tunability of the mechanical properties as well as their rapid healing capabilities potentiate these materials to find applications as remotely healable cell capture and release systems, or as cytocompatible soft fillers of biological cavities in tissue engineering and cell therapeutics. Therefore, we anticipate that our proposed materials will further “fuel” the field towards the development of even “smarter” self-repairing materials for biomedical applications and conceptualise their use in the clinical setting.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Leverhulme Trust (ECF-2013-472), the UCL Excellence Fellowship program and the Engineering and Physical Sciences Research Council (EPSRC, EP/M014649/1). We also thank Mr Bernardo Dias Lopes for his technical assistance in the preparation of the gels.Notes and references
- K. D. Birnbaum and A. S. Alvarado, Cell, 2008, 132, 697–710 CrossRef CAS PubMed.
- D. Y. Wu, S. Meure and D. Solomon, Prog. Polym. Sci., 2008, 33, 479–522 CrossRef CAS.
- S. Burattini, B. W. Greenland, D. Chappell, H. M. Colquhoun and W. Hayes, Chem. Soc. Rev., 2010, 39, 1973–1985 RSC.
- Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446–7467 RSC.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS PubMed.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS PubMed.
- C. E. Diesendruck, N. R. Sottos, J. S. Moore and S. R. White, Angew. Chem., Int. Ed., 2015, 54, 10428–10447 CrossRef CAS PubMed.
- P. Zhang and G. Li, Prog. Polym. Sci., 2016, 57, 32–63 CrossRef CAS.
- P. Cordier, F. Tournilhac, C. Soulie-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS PubMed.
- A. Phadke, C. Zhang, B. Arman, C.-C. Hsu, R. A. Mashelkar, A. K. Lele, M. J. Tauber, G. Arya and S. Varghese, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4383–4388 CrossRef CAS PubMed.
- J. Cui and A. d. Campo, Chem. Commun., 2012, 48, 9302–9304 RSC.
- S. Burattini, H. M. Colquhoun, J. D. Fox, D. Friedmann, B. W. Greenland, P. J. F. Harris, W. Hayes, M. E. Mackay and S. J. Rowan, Chem. Commun., 2009, 6717–6719 RSC.
- Y. Xu, Q. Wu, Y. Sun, H. Bai and G. Shi, ACS Nano, 2010, 4, 7358–7362 CrossRef CAS PubMed.
- Y.-G. Jia and X. X. Zhu, Chem. Mater., 2015, 27, 387–393 CrossRef CAS.
- E. A. Appel, F. Biedermann, U. Rauwald, S. T. Jones, J. M. Zayed and O. A. Scherman, J. Am. Chem. Soc., 2010, 132, 14251–14260 CrossRef CAS PubMed.
- T. Kakuta, Y. Takashima, M. Nakahata, M. Otsubo, H. Yamaguchi and A. Harada, Adv. Mater., 2013, 25, 2849–2853 CrossRef CAS PubMed.
- L. Peng, H. Zhang, A. Feng, M. Huo, Z. Wang, J. Hu, W. Gao and J. Yuan, Polym. Chem., 2015, 6, 3652–3659 RSC.
- Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339–343 CrossRef CAS PubMed.
- Y. Cao, T. G. Morrissey, E. Acome, S. I. Allec, B. M. Wong, C. Keplinger and C. Wang, Adv. Mater., 2017, 29, 1605099 CrossRef PubMed –n/a.
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334–337 CrossRef CAS PubMed.
- N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. C. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655 CrossRef CAS PubMed.
- S. Hou and P. X. Ma, Chem. Mater., 2015, 27, 7627–7635 CrossRef CAS PubMed.
- N. Kuhl, S. Bode, R. K. Bose, J. Vitz, A. Seifert, S. Hoeppener, S. J. Garcia, S. Spange, S. van der Zwaag, M. D. Hager and U. S. Schubert, Adv. Funct. Mater., 2015, 25, 3295–3301 CrossRef CAS.
- R. Chang, X. Wang, X. Li, H. An and J. Qin, ACS Appl. Mater. Interfaces, 2016, 8, 25544–25551 CAS.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- E. B. Murphy, E. Bolanos, C. Schaffner-Hamann, F. Wudl, S. R. Nutt and M. L. Auad, Macromolecules, 2008, 41, 5203–5209 CrossRef CAS.
- K. K. Oehlenschlaeger, J. O. Mueller, J. Brandt, S. Hilf, A. Lederer, M. Wilhelm, R. Graf, M. L. Coote, F. G. Schmidt and C. Barner-Kowollik, Adv. Mater., 2014, 26, 3561–3566 CrossRef CAS PubMed.
- G. B. Lyon, A. Baranek and C. N. Bowman, Adv. Funct. Mater., 2016, 26, 1477–1485 CrossRef CAS.
- J. Canadell, H. Goossens and B. Klumperman, Macromolecules, 2011, 44, 2536–2541 CrossRef CAS.
- J. A. Yoon, J. Kamada, K. Koynov, J. Mohin, R. Nicolaÿ, Y. Zhang, A. C. Balazs, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2012, 45, 142–149 CrossRef CAS.
- Z. Q. Lei, H. P. Xiang, Y. J. Yuan, M. Z. Rong and M. Q. Zhang, Chem. Mater., 2014, 26, 2038–2046 CrossRef CAS.
- E. B. Murphy and F. Wudl, Prog. Polym. Sci., 2010, 35, 223–251 CrossRef CAS.
- A. J. R. Amaral and G. Pasparakis, Polym. Chem., 2017, 8, 6464–6484 RSC.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- M. Motornov, Y. Roiter, I. Tokarev and S. Minko, Prog. Polym. Sci., 2010, 35, 174–211 CrossRef CAS.
- G. Pasparakis and M. Vamvakaki, Polym. Chem., 2011, 2, 1234–1248 RSC.
- Y. Zhang, B. Yang, X. Zhang, L. Xu, L. Tao, S. Li and Y. Wei, Chem. Commun., 2012, 48, 9305–9307 RSC.
- N. Hohlbein, A. Shaaban and A. M. Schmidt, Polymer, 2015, 69, 301–309 CrossRef CAS.
- H. Zhang and Y. Zhao, ACS Appl. Mater. Interfaces, 2013, 5, 13069–13075 CAS.
- R. Vaiyapuri, B. W. Greenland, H. M. Colquhoun, J. M. Elliott and W. Hayes, Polym. Chem., 2013, 4, 4902–4909 RSC.
- L. Chen, L. Si, F. Wu, S. Y. Chan, P. Yu and B. Fei, J. Mater. Chem. C, 2016, 4, 10018–10025 RSC.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- A. M. Alkilany and C. J. Murphy, J. Nanopart. Res., 2010, 12, 2313–2333 CrossRef CAS PubMed.
- J. Raeburn and D. J. Adams, Chem. Commun., 2015, 51, 5170–5180 RSC.
- N. Zhong and W. Post, Composites, Part A, 2015, 69, 226–239 CrossRef CAS.
- G. Springsteen and B. Wang, Tetrahedron, 2002, 58, 5291–5300 CrossRef CAS.
- Y. Guan and Y. Zhang, Chem. Soc. Rev., 2013, 42, 8106–8121 RSC.
- W. L. A. Brooks and B. S. Sumerlin, Chem. Rev., 2016, 116, 1375–1397 CrossRef CAS PubMed.
- S. D. Bull, M. G. Davidson, J. M. H. van den Elsen, J. S. Fossey, A. T. A. Jenkins, Y.-B. Jiang, Y. Kubo, F. Marken, K. Sakurai, J. Zhao and T. D. James, Acc. Chem. Res., 2013, 46, 312–326 CrossRef CAS PubMed.
- G. Vancoillie and R. Hoogenboom, Polym. Chem., 2016, 7, 5484–5495 RSC.
- L. He, D. E. Fullenkamp, J. G. Rivera and P. B. Messersmith, Chem. Commun., 2011, 47, 7497–7499 RSC.
- M. Piest, X. Zhang, J. Trinidad and J. F. J. Engbersen, Soft Matter, 2011, 7, 11111–11118 RSC.
- M. Nakahata, S. Mori, Y. Takashima, A. Hashidzume, H. Yamaguchi and A. Harada, ACS Macro Lett., 2014, 3, 337–340 CrossRef CAS.
- J. Collins, M. Nadgorny, Z. Xiao and L. A. Connal, Macromol. Rapid Commun., 2017, 38, 1600760 CrossRef PubMed.
- C. Zhang, M. D. Losego and P. V. Braun, Chem. Mater., 2013, 25, 3239–3250 CrossRef CAS.
- H. Meng, J. Zheng, X. Wen, Z. Cai, J. Zhang and T. Chen, Macromol. Rapid Commun., 2015, 36, 533–537 CrossRef CAS PubMed.
- S. Kitano, Y. Koyama, K. Kataoka, T. Okano and Y. Sakurai, J. Controlled Release, 1992, 19, 161–170 CrossRef CAS.
- T. Konno and K. Ishihara, Biomaterials, 2007, 28, 1770–1777 CrossRef CAS PubMed.
- A. Matsumoto, K. Kataoka and Y. Miyahara, Polym. J., 2014, 46, 483–491 CrossRef CAS.
- A. Pettignano, S. Grijalvo, M. Haring, R. Eritja, N. Tanchoux, F. Quignard and D. Diaz Diaz, Chem. Commun., 2017, 53, 3350–3353 RSC.
- O. R. Cromwell, J. Chung and Z. Guan, J. Am. Chem. Soc., 2015, 137, 6492–6495 CrossRef CAS PubMed.
- J. J. Cash, T. Kubo, A. P. Bapat and B. S. Sumerlin, Macromolecules, 2015, 48, 2098–2106 CrossRef CAS.
- M. Vatankhah-Varnoosfaderani, S. Hashmi, A. GhavamiNejad and F. J. Stadler, Polym. Chem., 2014, 5, 512–523 RSC.
- M. Shan, C. Gong, B. Li and G. Wu, Polym. Chem., 2017, 8, 2997–3005 RSC.
- G. Pasparakis, M. Vamvakaki, N. Krasnogor and C. Alexander, Soft Matter, 2009, 5, 3839–3841 RSC.
- A. J. R. Amaral and G. Pasparakis, Chem. Commun., 2015, 51, 17556–17559 RSC.
- A. J. R. Amaral and G. Pasparakis, ACS Appl. Mater. Interfaces, 2016, 8, 22930–22941 CAS.
- H. Zhang, D. Fortin, H. Xia and Y. Zhao, Macromol. Rapid Commun., 2013, 34, 1742–1746 CrossRef CAS PubMed.
- K. Bott, Z. Upton, K. Schrobback, M. Ehrbar, J. A. Hubbell, M. P. Lutolf and S. C. Rizzi, Biomaterials, 2010, 31, 8454–8464 CrossRef CAS PubMed.
- M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103, 655–663 CrossRef CAS PubMed.
- S. C. Rizzi, M. Ehrbar, S. Halstenberg, G. P. Raeber, H. G. Schmoekel, H. Hagenmüller, R. Müller, F. E. Weber and J. A. Hubbell, Biomacromolecules, 2006, 7, 3019–3029 CrossRef CAS PubMed.
- M. P. Lutolf, J. L. Lauer-Fields, H. G. Schmoekel, A. T. Metters, F. E. Weber, G. B. Fields and J. A. Hubbell, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5413–5418 CrossRef CAS PubMed.
- J. Chen, Q. Su, R. Guo, J. Zhang, A. Dong, C. Lin and J. Zhang, Macromol. Chem. Phys., 2017, 218, 1700166 CrossRef.
- C. E. Hoppe, M. Lazzari, I. Pardiñas-Blanco and M. A. López-Quintela, Langmuir, 2006, 22, 7027–7034 CrossRef CAS PubMed.
- ISO 10993-5/Ed.3, Biological evaluation of medical devices - Part 5: Tests for in vitro cytotoxicity, International Organization for Standardization, 2009 Search PubMed.
- V. Yesilyurt, M. J. Webber, E. A. Appel, C. Godwin, R. Langer and D. G. Anderson, Adv. Mater., 2016, 28, 86–91 CrossRef CAS PubMed.
- Z. Shen, K. Terao, Y. Maki, T. Dobashi, G. Ma and T. Yamamoto, Colloid. Polym. Sci., 2006, 284, 1001–1007 CAS.
- T. Aoki, Y. Nagao, K. Sanui, N. Ogata, A. Kikuchi, Y. Sakurai, K. Kataoka and T. Okano, Polym. J., 1996, 28, 371–374 CrossRef CAS.
- L. D. Taylor and L. D. Cerankowski, J. Polym. Sci., Polym. Chem. Ed., 1975, 13, 2551–2570 CrossRef CAS.
- K. Shiomori, A. E. Ivanov, I. Y. Galaev, Y. Kawano and B. Mattiasson, Macromol. Chem. Phys., 2004, 205, 27–34 CrossRef CAS.
- K. Kataoka, H. Miyazaki, T. Okano and Y. Sakurai, Macromolecules, 1994, 27, 1061–1062 CrossRef CAS.
- A. Matsumoto, T. Kurata, D. Shiino and K. Kataoka, Macromolecules, 2004, 37, 1502–1510 CrossRef CAS.
- H. Kaczmarek, A. Kamińska, M. Światek and J. F. Rabek, Angew. Makromol. Chem., 1998, 261–262, 109–121 CrossRef CAS.
- A. E. Ivanov, H. Larsson, I. Y. Galaev and B. Mattiasson, Polymer, 2004, 45, 2495–2505 CrossRef CAS.
- H. Liu, Y. Li, K. Sun, J. Fan, P. Zhang, J. Meng, S. Wang and L. Jiang, J. Am. Chem. Soc., 2013, 135, 7603–7609 CrossRef CAS PubMed.
- S. K. Ghosh and T. Pal, Chem. Rev., 2007, 107, 4797–4862 CrossRef CAS PubMed.
- S. H. Brewer, A. M. Allen, S. E. Lappi, T. L. Chasse, K. A. Briggman, C. B. Gorman and S. Franzen, Langmuir, 2004, 20, 5512–5520 CrossRef CAS PubMed.
- Z. Tang, Y. Guan and Y. Zhang, Polym. Chem., 2014, 5, 1782–1790 RSC.
- H. Ge, Y. Ding, C. Ma and G. Zhang, J. Phys. Chem. B, 2006, 110, 20635–20639 CrossRef CAS PubMed.
- G. Carré de Lusançay, S. Norvez and I. Iliopoulos, Eur. Polym. J., 2010, 46, 1367–1373 CrossRef.
- X. Xue, G. Pasparakis, N. Halliday, K. Winzer, S. M. Howdle, C. J. Cramphorn, N. R. Cameron, P. M. Gardner, B. G. Davis, F. Fernández-Trillo and C. Alexander, Angew. Chem., Int. Ed., 2011, 50, 9852–9856 CrossRef CAS PubMed.
- G. Springsteen and B. Wang, Chem. Commun., 2001, 1608–1609 RSC.
- A. G. Skirtach, C. Dejugnat, D. Braun, A. S. Susha, A. L. Rogach, W. J. Parak, H. Möhwald and G. B. Sukhorukov, Nano Lett., 2005, 5, 1371–1377 CrossRef CAS PubMed.
- M. C. Roberts, M. C. Hanson, A. P. Massey, E. A. Karren and P. F. Kiser, Adv. Mater., 2007, 19, 2503–2507 CrossRef CAS.
- D. D. McKinnon, D. W. Domaille, J. N. Cha and K. S. Anseth, Adv. Mater., 2014, 26, 865–872 CrossRef CAS PubMed.
- M. W. Conway, S. W. Almond, J. E. Briscoe and L. E. Harris, J. Pet. Technol., 1983, 35, 315–320 CrossRef CAS.
- F. van de Manakker, M. van der Pot, T. Vermonden, C. F. van Nostrum and W. E. Hennink, Macromolecules, 2008, 41, 1766–1773 CrossRef CAS.
- L. Wang, M. Liu, C. Gao, L. Ma and D. Cui, React. Funct. Polym., 2010, 70, 159–167 CrossRef CAS.
- I. Cobo, M. Li, B. S. Sumerlin and S. Perrier, Nat. Mater., 2015, 14, 143–159 CrossRef CAS PubMed.
- B.-H. Lee, B. Li and S. A. Guelcher, Acta Biomater., 2012, 8, 1693–1702 CrossRef CAS PubMed.
- T. Canal and N. A. Peppas, J. Biomed. Mater. Res., 1989, 23, 1183–1193 CrossRef CAS PubMed.
- F. R. Maia, K. B. Fonseca, G. Rodrigues, P. L. Granja and C. C. Barrias, Acta Biomater., 2014, 10, 3197–3208 CrossRef CAS PubMed.
- Q. Wen, A. Basu, P. A. Janmey and A. G. Yodh, Soft Matter, 2012, 8, 8039–8049 RSC.
- G. P. Raeber, M. P. Lutolf and J. A. Hubbell, Biophys. J., 2005, 89, 1374–1388 CrossRef CAS PubMed.
- G. Deng, F. Li, H. Yu, F. Liu, C. Liu, W. Sun, H. Jiang and Y. Chen, ACS Macro Lett., 2012, 1, 275–279 CrossRef CAS.
- Z. Wei, J. H. Yang, J. Zhou, F. Xu, M. Zrinyi, P. H. Dussault, Y. Osada and Y. M. Chen, Chem. Soc. Rev., 2014, 43, 8114–8131 RSC.
- D. L. Taylor and M. in het Panhuis, Adv. Mater., 2016, 28, 9060–9093 CrossRef CAS PubMed.
- A. Banerjee, M. Arha, S. Choudhary, R. S. Ashton, S. R. Bhatia, D. V. Schaffer and R. S. Kane, Biomaterials, 2009, 30, 4695–4699 CrossRef CAS PubMed.
- H. Oda, T. Konno and K. Ishihara, Biomaterials, 2013, 34, 5891–5896 CrossRef CAS PubMed.
- L. Cai, R. E. Dewi and S. C. Heilshorn, Adv. Funct. Mater., 2015, 25, 1344–1351 CrossRef CAS PubMed.
- H. Wang and S. C. Heilshorn, Adv. Mater., 2015, 27, 3717–3736 CrossRef CAS PubMed.
- X. Yang, G. Liu, L. Peng, J. Guo, L. Tao, J. Yuan, C. Chang, Y. Wei and L. Zhang, Adv. Funct. Mater., 2017, 27, 1703174 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental procedures and data. See DOI: 10.1039/c7py01202k |
| This journal is © The Royal Society of Chemistry 2018 |