
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA photochemical approach for evaluating the reactivity of substituted lappaconitines
Aleksandra A.
Ageeva
 *a,
Ekaterina A.
Khramtsova
ab,
Viktor F.
Plyusnin
ab,
Aleksandr A.
Stepanov
a and
Tatyana V.
Leshina
a
*a,
Ekaterina A.
Khramtsova
ab,
Viktor F.
Plyusnin
ab,
Aleksandr A.
Stepanov
a and
Tatyana V.
Leshina
a
aVoevodsky Institute of Chemical Kinetics and Combustion SB RAS, Institutskaya st., 3, 630090 Novosibirsk, Russia. E-mail: al.ageeva@gmail.com
bNovosibirsk State University, Pirogova st., 2, 630090 Novosibirsk, Russia
First published on 4th December 2017
Abstract
Lappaconitine (LC) is a natural diterpenoid alkaloid (DTA), acting as a human heart sodium channel blocker and possessing a wide range of biological activities, the cellular and molecular mechanisms of which are widely studied. This interest is due to the fact that various representatives of this DTA class show opposite biological activities. The possible reasons for this difference seem to be related to the peculiarities of the substituent effect on the drug–receptor binding process. In this work, the influence of substituents on the reactivity of LC and its derivatives has been studied by using elementary processes of photodecomposition. The given approach includes the joint analysis of the photophysical properties of the studied systems and their photodecomposition quantum yields. It allows us to trace the influence of substituents, located in the diterpenoid skeleton and anthranilic fragment, on processes in both moieties of LC. Summarizing the data obtained, an inverse dependence of fluorescence and photodegradation quantum yields has been observed. This correlation established for LCs, in particular, allows one to propose a way to evaluate the photostability of potential drugs based on fluorescence analysis. This would be appropriate for compounds in which the reactivity depends on intersystem crossing, i.e. in the cases where the initial and reacting excited states differ in multiplicity.
Introduction
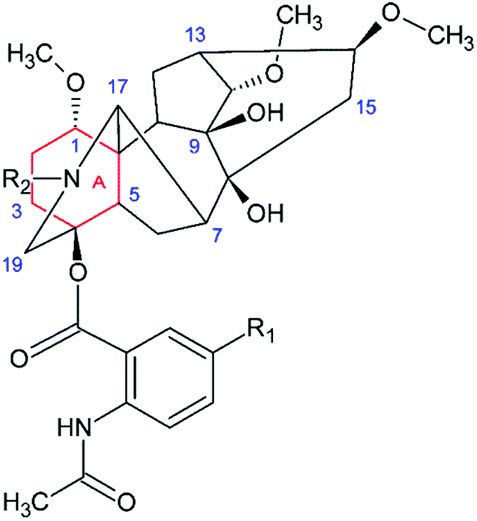
Lappaconitine (LC) is a natural diterpenoid alkaloid (DTA) that possesses a wide range of biological activities, including antiarrhythmic,1 anti-inflammatory,1–3 antioxidative,3 anticancer,4 epileptiform5,6etc. It is widely used for pain-control management in China. Furthermore, lappaconitine hydrobromide (Allapinin) represents a class of antiarrhythmic drugs in the Russian Federation.7 DTAs as well as LC, isolated from the plants of genera Aconitum, comprise a large group of antiarrhythmic and arrhythmogenic agents.8 So, one of the arrhythmogenic agents, aconitine, a site 2 sodium channel neurotoxin, induces severe neurological symptoms and cardiovascular collapse.9 This is even more interesting because studies at the cellular level have shown that LC and aconitine bind to the sodium channels in a similar way. Nevertheless, the binding mechanism of both toxins has not yet been established. However, it is supposed that LC can bind to the open sodium channels by several ways, including penetrating the membranes or directly into the pores, and blocking the receptors by the allosteric mechanism. And studies of the cellular mechanisms of LC and aconitine action have shown that they bind to the same or similar receptors.9 This binding is considered to occur with the participation of diterpenoid skeletons, identical for LC and aconitine.9 In view of these facts, the researchers arrived at a conclusion that differences should be sought in the influence of the substituents located in the diterpenoid skeleton.9 Attempts to trace the “structure–property” relationship of a few DTAs have been made in a review.8 The principal problems in establishing any regularity arise from the fact that DTAs, both antiarrhythmic and arrhythmogenic, contain almost the same set of substituents, viz., there are one ethyl group at the nitrogen atom in the DTA fragment, several hydroxy and methoxy groups, and one aromatic substituent. The latter is the fragment of benzoic or anthranilic acid (AA). Antiarrhythmic and arrhythmogenic DTAs mainly differ from each other in the arrangement of these substituents. Thereby LC and the strongest toxin – aconitine – have different aromatic substituents with different localization. So, the N-acetyl-AA fragment of LC is located at the 4 position, whereas the benzoyl residue of aconitine is at the 14 position (Fig. 1).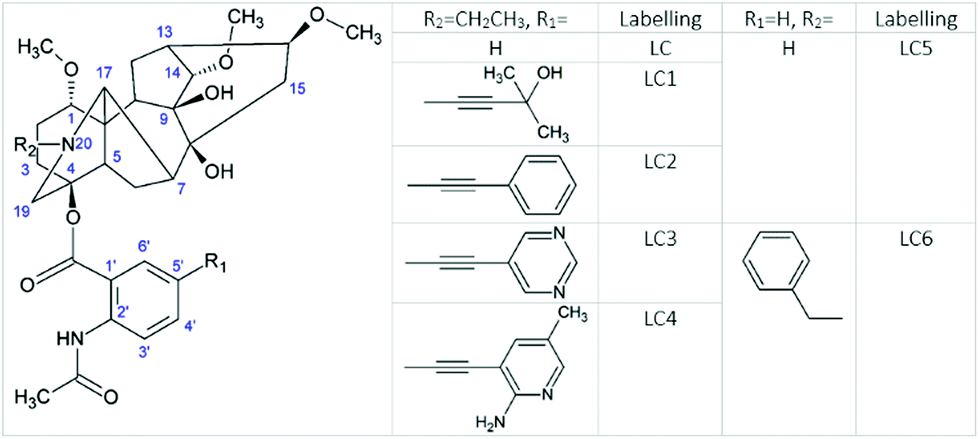 | ||
| Fig. 1 The molecular structure of LC (R1 = H, R2 = CH2CH3) and its derivatives. | ||
In view of this, the aromatic substituent plays a significant role in the therapeutic activity of DTA. In this report, we made an effort to establish the structure–property relationship by the example of substituted (not natural) LCs, varying the acceptor properties of the aromatic fragment. Furthermore, we will study the impact of the steric and electronic effects of substituents on the elementary process of intramolecular electron transfer (ET) between diterpenoid and N-acetyl-AA moieties. Moreover, photoinduced ET has been successfully used in several studies to aid in understanding the peculiarities of biological processes, including drug–receptor binding.10–12
The use of ET for modelling biological processes is advantageous when the processes of drug–receptor binding include charge redistribution. For instance, it might be the formation of hydrogen bonds and complexes with charge transfer.12 In the case that a DTA binds to the sodium receptors via the diterpenoid fragment according to ref. 9, the donor ability of the nitrogen atom in the 20 position will play the main role. If, as it has been supposed in ref. 10, the binding process involves the charge transfer between the AA moiety and amino acid residues located in receptor active sites, the acceptor ability of the AA moiety will be the main factor.
Therefore, we expect that the effects of substituents on the ET between N(20) and AA fragments of modified LCs to some extent will be useful for understanding the impact of substituents on LC binding. Although substituted LCs in this study (see Fig. 1) have rather different substituents from natural DTAs, one can assume that the same steric and electronic effects will determine their influence on the binding mechanism. The given approach, including the study of ET, allows one to trace the effect of substituents on both parts of DTA molecules. It is noteworthy that for the modified LCs from Fig. 1, a detailed mechanism of photolysis has been established by CIDNP and EPR analysis in previous studies.10,13–16 According to these studies, a photoreaction begins from the partially reversible intramolecular electron transfer (ET) between nitrogen located in the diterpenoid skeleton and the AA fragment with a subsequent cleavage of the ether bond. Since the anthranilic fragment is a chromophore in these molecules as well as a potential electron acceptor in redox processes, substituents have a significant impact on the efficiency of ET and subsequent photodegradation, respectively. It has been suggested that the photocleavage of LC derivatives depends on the proportion between singlet and triplet excited states.16 To understand the nature of the influence of substituents and their impact on the formation of singlet and triplet excited states, this report is focused on the examination of the photophysical properties of LC and its substituted analogues and their photodecomposition quantum yields.
It should be noted that, in contrast to LC, the photophysical properties of many other ethers of anthranilic acid (AA), to which LC relates, are broadly investigated.17–22 There are methyl ethers of AA (methyl anthranilate, MA),17,21,22 menthyl ethers of AA18 and N-acetyl-menthylanthranilate.19 In this regard, and because the anthranilic part of lappaconitine is a chromophore in this molecule, in this paper we will compare the photophysical properties of LC and its derivatives with those of MA. This work includes the investigation of fluorescence lifetimes and fluorescence and photodecomposition quantum yields. In addition, we have analysed the products of phototransformation by 1H-NMR. The final purpose of this study is to establish the “structure–property” relationship by studying the correlation of the photophysical properties (fluorescence lifetimes and quantum yields) of LC and its derivatives with the photodegradation effectivity of these compounds. In particular, we are going to trace the contribution to the reactivity from singlet and triplet excited states and the impact of the substitution on the ratio of these states. The advantages of this approach are that the characteristics of the chromophore fluorescence will reflect the influence of all substituents, including those located in the diterpenoid skeleton.
Experimental
Materials
LC was isolated from the root of Aconitum septentrionale Koelle in accordance with the known procedure23 before using an additional purification procedure by recrystallization from methanol. The synthesis of substituted LC was described in ref. 24 and 25; the compound was purified by recrystallization from methanol. Methanol and deuteromethanol (Aldrich, D 99.8%) were used as solvents. The typical LC concentration was about 10−5 M.Instrumentation
All UV spectroscopic measurements were performed using a quartz cuvette of 1 cm optical length. Methanol was used as a solvent.Spectra and kinetic curves of luminescence were recorded with an Edinburgh Instruments FLSP-920 spectrofluorimeter with either a Xenon lamp or a laser diode EPLED-300 (λex = 300 nm, pulse duration 0.6 ns) as excitation sources. Each sample was bubbled with argon for 30 min to remove dissolved oxygen just before the experiments. The absorption spectra were recorded using an Agilent 8453 spectrophotometer.
The steady state photolysis experiments of LC in deoxygenized solutions were performed using an excimer lamp (λ = 308 nm) as a light source. The intensity of light was measured by using a SOLO-2 “Gentec” power meter. The process of photolysis was monitored using UV and 1H-NMR spectroscopy.1H-NMR experiments were performed with a Bruker AVHD (500 MHz 1H operating frequency, τ (90) = 11.2 μs) NMR spectrometer.
Methods
Fluorescence quantum yields (φ) were determined by integrating the corrected emission spectra and using a solution of anthracene in methanol (φ = 0.27) as a standard.18 For the determination of φ the calculation using the following equation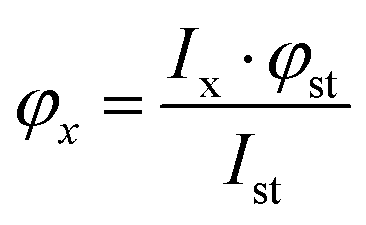 | (1) |
The photodecomposition quantum yield of LC was estimated using changes in absorbance from the time of irradiation. Absorbance data were fitted to an exponential decay of the form
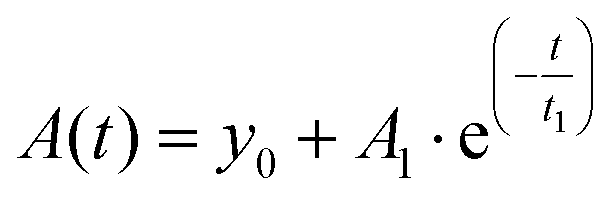 | (2) |
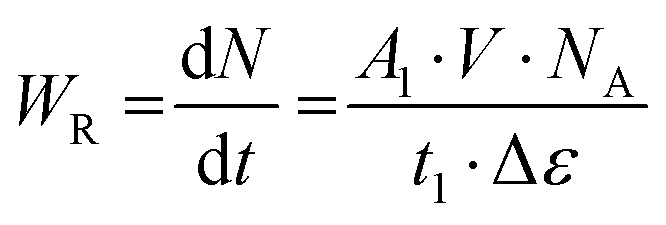 | (3) |
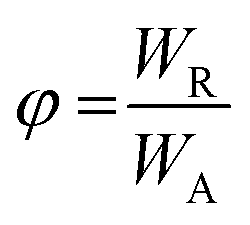 | (4) |
 | (5) |
Simultaneously, the process of LC photodecomposition was monitored by 1H-NMR. The aliquots were taken before UV irradiation and after 5, 10, and 23 minutes of irradiation with an excimer lamp. In order to verify the photodecomposition quantum yields, calculated from the absorbance data, we analysed the NMR signals of the initial LC and its photoproducts and calculated the quantum yields from the NMR data. The changes of LC concentration (proportional to the integral intensity of NMR signals) were fitted to an exponential decay of the form mentioned above. The parameter y0 was fixed and equalled to zero since the signals of the initial LC were observed completely decaying during the photoreaction. The comparative analysis of quantum yields has shown an agreement within 10%.
Results
Absorption spectra
The UV absorption spectra of MA, LC and its derivatives in methanol are shown in Fig. 2. As can be seen in Fig. 2, the blue shift of the LC long-wave absorption band compared with MA is observed in the UV spectrum. In the literature26 the same effect has been studied, the UV absorption spectra of N-acetyl-AA relative to the spectrum of AA, and it has been described and interpreted in terms of the electronic and steric impacts of the substituents.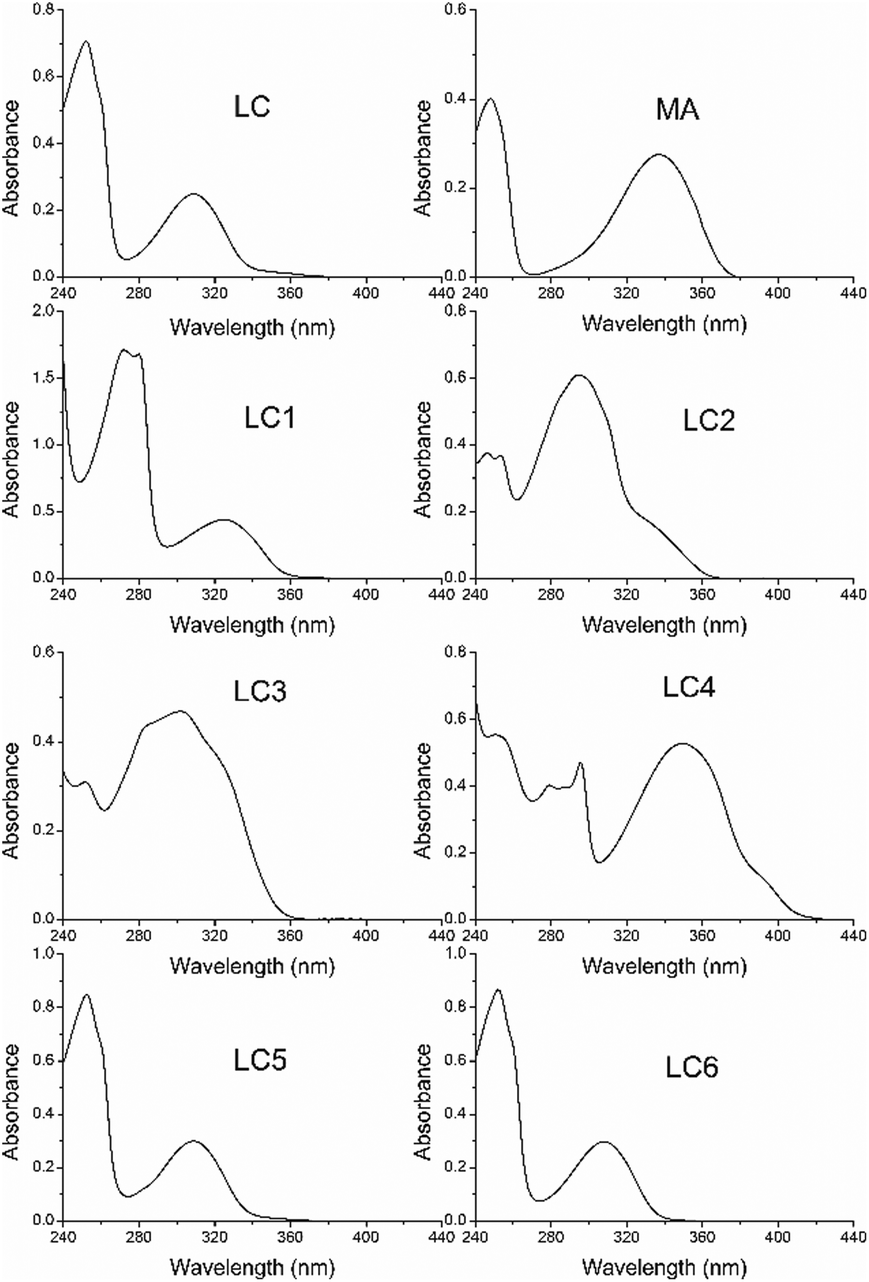 | ||
| Fig. 2 The UV-visible absorption spectra of LC (6.4 × 10−5 M), MA (5.1 × 10−5 M) and LC derivatives (∼10−5 M) in methanol in a 1 cm quartz cuvette. | ||
The blue shift and reduction in the intensity of the long-wave band relative to the spectrum of AA have been attributed to the increase in conjugation of the system following acylation of the amine group and reduced conjugation between the nitrogen atom and the aromatic ring arising from the loss of planarity of the molecule, respectively. The extinction coefficients of LC and its derivatives are presented in Table 1.
| Compound | ε at λ = 308 nm, M−1 cm−1 |
|---|---|
| MA | 2100 ± 100 |
| LC | 3900 ± 100 |
| LC1 | 1900 ± 200 |
| LC2 | 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ± 1000 000 ± 1000 |
| LC3 | 13700 ± 700 |
| LC4 | 6400 ± 300 |
| LC5 | 3900 ± 200 |
| LC6 | 3900 ± 200 |
Steady state photolysis
As shown in Fig. 3, the UV light irradiation at 308 nm of LC generates spectral changes corresponding to the photo-cleavage. | ||
| Fig. 3 Absorption spectra of LC (8 × 10−5 M) photodecomposition in methanol upon irradiation at 308 nm and a light intensity of 2.8 mW cm−2 after various irradiation times. Inset: Changes of absorbance at 308 nm; blue plots – experimental data; red line – fitting to the first order exponential decay. | ||
We observe from Fig. 3 that there is not only one process in the photolysis of LC (see Fig. 4). Approximately, after 20 min of irradiation with an excimer lamp, the isosbestic point at 298 nm disappears and the second phototransformation process begins. To understand what processes occur in the given system under UV-irradiation, we studied the products of photolysis by NMR spectroscopy.
 | ||
| Fig. 4 Molecular structure of photo-Fries rearrangement photoproducts, where R1 is acetyl and R2 is methyl. | ||
1H-NMR spectra
The products of LC photodecomposition can be identified from Fig. 5. In Fig. 5, we compare the NMR-spectra of aromatic areas for AA (green), initial LC (blue) and a mixture of LC and its photoproducts (red). Comparing the NMR-spectra of LC and AA we conclude that LC phototransformation involves the accumulation of the main reaction product N-acetyl-AA, and its further photolysis through the deacylation with the formation of AA. The signals, in the region of the aromatic proton resonance, two doublets located between 7.70 and 8.00 ppm and a singlet at 8.40 ppm, can be presumably attributed to the products of the photo-Fries rearrangement (5-methyl-2-aminobenzoic acid and 5-aceto-2-aminobenzoic acid (see Fig. 4)). Two doublets located at 7.26 and 7.00 ppm might belong to 3-aceto-2-aminobenzoic acid or 3-methyl-2-aminobenzoic acid (Fig. 4). Furthermore, these doublets may be ascribed to the compounds formed in reactions 5.2 or 5.1 (a full scheme is presented in Fig. 7), if one supposes that two missing triplets are overlapped with the signals of the main products or the initial compound (7.20–7.30 ppm, 7.50–7.60 ppm).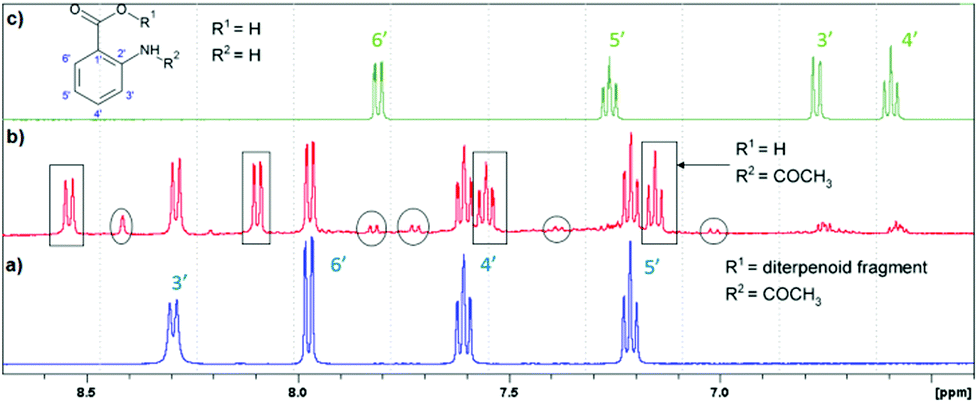 | ||
| Fig. 5 1H-NMR spectra of LC (a, blue), a mixture of LC and its photoproducts (b, red) after 120 min of irradiation at 308 nm (in rectangles – the main photoproduct N-acetyl-AA; in circles – the photo-Fries rearrangement products), and AA (c, green) in methanol. | ||
Thus, according to the analysis of the aromatic region of the NMR spectra of the reaction mixture, it contains the following photoproducts: LC as the starting material, N-acetyl-AA, AA and presumably the photo-Fries rearrangement products. The products of the diterpenoid fragment transformation coincide with those identified earlier for LC.14
Fluorescence measurements
The emission spectra of LC and MA in methanol are shown in Fig. 6 and demonstrate emission maxima at 375 and 410 nm, respectively. The quantum yields and lifetimes are given in Table 2. | ||
| Fig. 6 The corrected fluorescence emission spectra of LC and MA in methanol, λex = 300 nm (top); fluorescence decay traces of LC and MA at 375 and 410 nm (λex = 300 nm); IRF – instrument response function (below). | ||
| Labelling | φ fl (λex = 300 nm) | Φ (λex = 308 nm) | τ fl, ns |
|---|---|---|---|
| MA | 0.59 ± 0.09 | — | 7.3 ± 0.7 |
| LC | 0.017 ± 0.006 | 0.19 ± 0.02 | 2.1 ± 0.2 |
| LC1 | 0.027 ± 0.04 | 0.16 ± 0.02 | 2.4 ± 0.2 |
| LC2 | 0.042 ± 0.006 | 0.056 ± 0.006 | 3.1 ± 0.3 |
| LC3 | 0.023 ± 0.004 | 0.15 ± 0.01 | 2.5 ± 0.3 |
| LC4 | 0.030 ± 0.005 (λex = 300) | 0.023 ± 0.002 | 3.2 ± 0.3 |
| 0.065 ± 0.01 (λex = 375) | 2.4 ± 0.2 | ||
| LC5 | 0.17 ± 0.03 | 0.48 ± 0.05 | 1.2 ± 0.1 |
| LC6 | 0.025 ± 0.004 | 0.35 ± 0.04 | 1.8 ± 0.2 |
A comparison of the fluorescence quantum yields and lifetimes of LC and MA has shown the appreciable reduction of these values for LC. The value of τfl for LC is three times less than that of MA and almost two times less than that of N-acetyl-MA. According to ref. 27, the high quantum yield of fluorescence and a red shift in the luminescence and absorption spectra of N-deacetyl LC compared to those of LC can be explained by an increase in the electron releasing ability of the –NRH group following the deacetylation. In addition, the shortening of the LC fluorescence lifetime (2.15 ns), in comparison with N-acetyl-AA (4 ns), is most likely to reveal the process of the fluorescence quenching of the anthranilic fragment through the previously described ET from the amine group of the alkaloid to the anthranilic fragment.10,13–16
In this connection, the difference between LC and MA fluorescence quantum yields and lifetimes is caused by two factors: the influence of the acetyl group leading to the loss of the molecular planarity and a decrease of the electron-donating character of the amine group towards the aromatic moiety and the process of intramolecular ET. Table 2 summarizes the fluorescence data obtained for all derivatives of LC.
As can be seen from the table, the introduction of additional substituents in both the anthranilic fragment and diterpenoid (the substitution of the ethyl group for the nitrogen atom by H and benzyl) does not lead to significant changes in the lifetime of fluorescence.
Discussion
The full scheme of LC transformation under UV-irradiation is presented in Fig. 7. The stages 1–4 and 6–7 have been established earlier by the analysis of spin effects,10,13–16 while steps 5.1–5.3 are proposed based on the analysis of the NMR spectra of photolysis products in this work. The photolysis of N-acetyl-AA is considered to involve a cleavage and photo-Fries rearrangement.20 So, it has been shown that the N-acetyl-AA solution after UV-irradiation becomes a mixture of the starting material, AA, 3-aceto-2-aminobenzoic acid, and 5-aceto-2-aminobenzoic acid, all products of cleavage and rearrangements. | ||
| Fig. 7 Full scheme of LC transformation under UV irradiation. | ||
Despite the fact that this is a minor process, this may be important because the photo-Fries rearrangement is accompanied by the formation of short-lived paramagnetic intermediates, leading to the formation of short-lived free toxic radicals. The latter are known to be the cause of damaging effects on cellular structures such as cell membranes or DNA.
The singlet-excited state of AA compounds is known to fluoresce, and undergo non-radiative deactivation or intersystem crossing with the formation of the reactive triplet excited state.18–20 Since the photodegradation of LC has been established previously occurring from the triplet-excited state, an attempt was made to evaluate the quantum yield of the LC triplet state using nanosecond laser flash photolysis. However, an extremely low triplet quantum yield was observed φT = 0.008. We attribute this to the fact that the rate constant of ET has to be substantially high (108–109 s−1), as follows from the manifestation of the initial LC CIDNP effect.10,13–16 It allows us to observe only a little effective yield of the triplet state.
Therefore, to trace the contribution of singlet and triplet excited states to LC photodegradation one can only compare fluorescence and photodecomposition quantum yields, taking into account the decomposition considered to occur through the triplet-excited state.10,13–16 According to the modern point of view, intersystem crossing between singlet and triplet excited states in compounds, involving closely located amine and carbonyl groups, might be caused by the vibrational interaction between close-lying S1(π,π*) and S2(n,π*) states.28 This interaction leads to a large increase in the non-radiative decay rate of the lowest excited singlet state. At the same time, intermolecular hydrogen bonds, changing the planarity of the nitrogen atom, conversely result in level stabilization and intersystem crossing to a T1(n,π*) triplet state.28 The influence of intra- and intermolecular hydrogen bonds on the LC photoinduced processes was proved in ref. 15. The authors observed decreases of LC photodegradation when it was solubilized in glycyrrhizic acid. This phenomenon was explained on account of the intramolecular hydrogen bond influence on the deactivation pathways of the LC singlet excited state. Hence, it can be seen that there are factors contributing to the effectivity of either the radiative or the non-radiative decay of the LC singlet excited state and the decrease of its internal conversion into the reactive triplet state.
We assume that the introduction of other substituents into the diterpenoid part of LC or in its anthranilic fragment can also affect the singlet–triplet crossing through the changing steric and electronic effects. Furthermore, we compared the fluorescence and photodecomposition quantum yields of LCs, in which the substituents are located in the anthranilic fragment. The result of this comparison is the inverse qualitative correlation between the quantum yield values of fluorescence and photodecomposition for various LCs (Fig. 8).
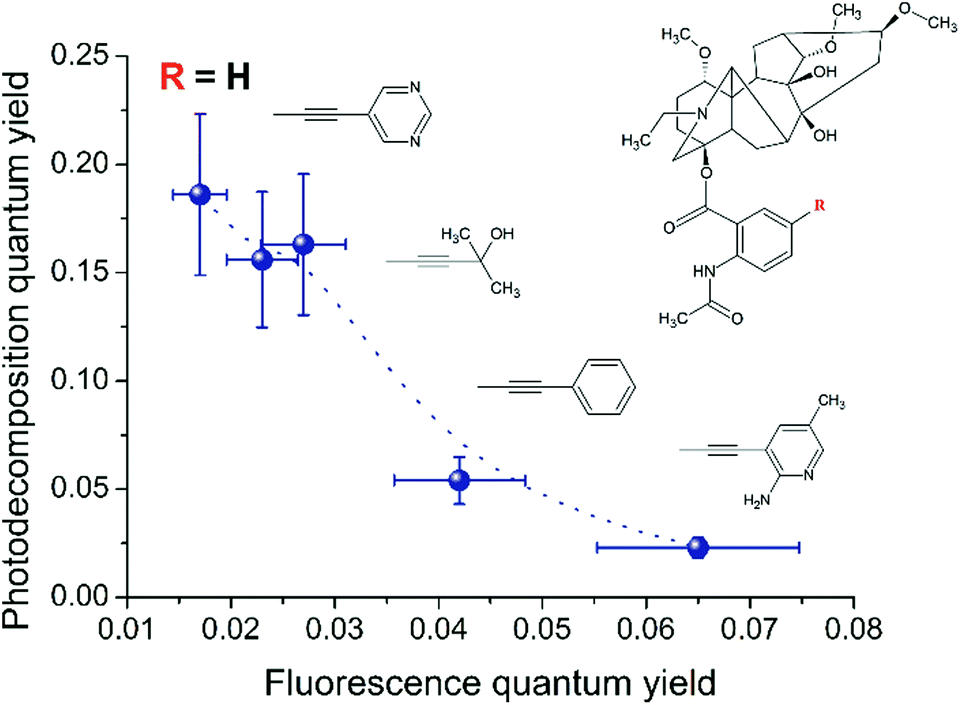 | ||
| Fig. 8 Correlation between the fluorescence and photodecomposition quantum yields of LC derivatives. | ||
At the same time, the data related to LC4 need some clarification. It was excited at two wavelengths (300 and 375 nm). The value of the fluorescence quantum yield obtained at 375 nm was used for correlation in connection with the following circumstances. Firstly, LC4 shows a minimal photodecomposition quantum yield (Table 2), and its absorption spectrum is noticeably shifted to the long-wavelength region by contrast to other derivatives (Fig. 2). Secondly, it is known from the study of substituted LC photolysis by the CIDNP technique that in the case of aromatic substituents containing functional groups, a substituent also participates as a partner in the ET.16 In the case of LC4, biradical zwitterions (BZ) are likely to be formed with positive centers both on the nitrogen of the diterpenoid skeleton and on the amino methyl pyridine (Fig. 9). If the amino methyl pyridine is involved in the ET process, then one can expect that the BZ, where the donor is separated from the acceptor by the acetylene bridge known to have high conductivity, will be more effective in the reversible ET. The back ET will lead to a decrease in the quantum yield of photodecomposition. In this case, it is reasonable to use for the correlation the value of the fluorescence quantum yield, measured at the wavelength of the excited substituent (375 nm).
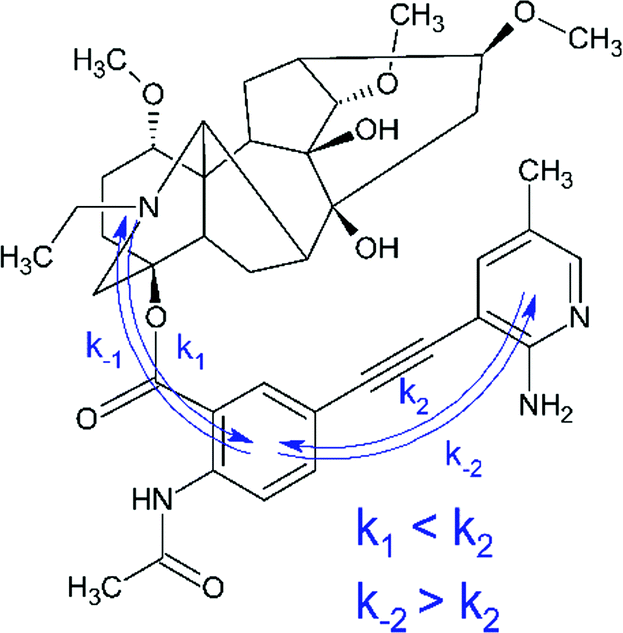 | ||
| Fig. 9 Two possible ET processes (subscripts 1 and 2) in LC4 leading to the formation of two BZ. | ||
Thus, the above correlation confirms the relationship between the photophysical properties and the reactivity of LC and its derivatives, assumed in ref. 16. Since the substituents in compounds LC1–LC4 have rather weak multidirectional effects and all of them include a rigid acetylene bridge, one can expect that they are likely to change the geometry of the molecule. Indeed, according to the reference data in LC hydrobromide salts, the nitrogen atom in the diterpenoid is protonated and forms an intramolecular H-bond with O–C1 causing the “boat” conformation of ring A (see Fig. 10).29 On the other hand, the absence of an intramolecular H-bond leads to the change of the conformation to the “chair”. Since the interaction of the drug with amino acid residues located in receptor active sites often involves the formation of hydrogen bonds, it is reasonably assumed that it would lead to a change in the conformation. Hence, the change of the binding conditions, depending on the location, for example –OH groups, takes place.9 It might be one of the reasons of the difference between the binding of antiarrhythmic and arrhythmogenic DTA agents.
 | ||
| Fig. 10 The ring A (red) adopted the boat or chair conformation depending on the presence or absence of an intramolecular H-bond between H–N and O–C1. | ||
At the same time, the changes of the fluorescence and photodegradation quantum yields of LC5 and LC6, where the substituents located at the nitrogen atom in the diterpenoid skeleton are altered from the ethyl group to hydrogen or benzyl, in comparison with LC, are very large. LC5 demonstrates extremely high fluorescence and photodecomposition quantum yields, while LC6 exhibits a high photodecomposition quantum yield.
In our opinion, this may mean not only a change of conformation, but also a change in the photoreaction mechanism due to electronic and steric factors. We assume that replacing of the ethyl group by –H and –CH2C6H5 will decrease the electron-donating ability of the diterpenoid's nitrogen atom. One can expect that this will reduce the efficiency of the ET process and the breakdown of the ether bond will occur through a concerted mechanism, i.e. without the formation of intermediate radical particles. A previous investigation of LC5–LC6 by the CIDNP technique has shown the CIDNP effects similar to those observed for LC and other derivatives.16 Since the CIDNP effects are known to reveal only the radical pathway of photoreaction, we can suggest for LC5 and LC6 the formation of most part of photoproducts via other mechanisms.
In addition, the data in Table 2 demonstrate that the variation of substituents in the diterpenoid fragment seems to lead to more significant changes of photodecomposition quantum yields than the similar variation in the AA fragment. It is associated with more significant alteration in geometry (steric factors) and electronic factors in comparison with the changes caused by the variation of the substituents in the chromophore–anthranilic fragment. The significant effect of substituents located in the alkaloid fragment of LC may be important since this fragment is known to be involved in the process of DTA binding with the receptors.
Conclusion
Thus, it can be stated that electronic effects as well as steric hindrance make a significant contribution to the photophysical properties and the reactivity of LC and its derivatives. The analysis of the fluorescence and photodecomposition quantum yields shows the influence of substituents located in both parts of the DTA, the diterpenoid skeleton and the anthranilic fragment, on the photophysical properties and reactivity of LCs. This work also traces the relationship between these characteristics. Meanwhile, based on the establishment of a correlation between the quantum yields of fluorescence and photodegradation for different LCs, one can propose an approach for evaluating the photostability of other potential drugs. This approach would be appropriate for compounds in which, similar to LCs, the reactivity depends on intersystem crossing, i.e. in the cases where the initial and reacting excited states have different multiplicity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the grant 16-33-00412 of the Russian Foundation of Basic Research.References
- J. F. Heubach and A. Schüle, Planta Med., 1998, 64, 22–26 CrossRef CAS PubMed.
- Y. Z. Wang, Y. Q. Xiao, C. Zhang and X. M. Sun, J. Tradit. Clin. Med., 2009, 29, 141–145 CrossRef.
- F. Shaheen, M. Ahmad, M. T. H. Khan, S. Jalil, A. Ejaz, M. N. Sultankhodjaev, M. Arfan, M. I. Choudhary and Atta-ur-Rahman, Phytochemistry, 2005, 66, 935–940 CrossRef CAS PubMed.
- G. I. Solyanik, A. G. Fedorchuk, O. N. Pyaskovskaya, O. I. Dasyukecitch, N. N. Khranovskaya, G. N. Aksenov and V. V. Sobetsky, Exp. Oncol., 2004, 26, 307–311 Search PubMed.
- A. Ameri, Brain Res., 1997, 767, 36–43 CrossRef.
- A. Ameri and T. Simmet, Brain Res., 1999, 842, 332–341 CrossRef CAS PubMed.
- Yu. N. Zamataev, Yu. A. Kremnev, N. A. Grinenko and S. E. Podshibyakin, Russ. Med. Zh. [Russ. Med. J.], 2003, 9, 518–521 Search PubMed (in Russian).
- M. S. Yunusov, Russ. Chem. Bull. Int. Ed., 2011, 60, 633–638 CrossRef CAS.
- S. N. Wright, Mol. Pharmacol., 2001, 59, 183–192 CAS.
- N. E. Polyakov, V. K. Khan, M. B. Taraban, T. V. Leshina, O. A. Luzina, N. F. Salakhutdinov and G. A. Tolstikov, Org. Biomol. Chem., 2005, 3, 881–885 CAS.
- F. I. Ataullahanov and A. M. Jabotinsky, Biophysics, 1975, 20, 596–601 Search PubMed.
- E. A. Khramtsova, D. V. Sosnovsky, A. A. Ageeva, E. Nuin, M. L. Marin, P. A. Purtov, S. S. Borisevich, S. L. Khursan, H. D. Roth, M. A. Miranda, V. F. Plyusnin and T. V. Leshina, Phys. Chem. Chem. Phys., 2016, 18, 12733–12741 RSC.
- N. E. Polyakov and T. V. Leshina, Russ. Chem. Bull. Int. Ed., 2007, 56, 631–642 CrossRef CAS.
- N. E. Polyakov, T. V. Leshina, A. V. Tkachev, I. A. Nikitina and N. A. Pankrushina, J. Photochem. Photobiol., A, 2008, 197, 290–294 CrossRef CAS.
- N. E. Polyakov, O. A. Simaeva, M. B. Taraban, T. V. Leshina, T. A. Konovalova, L. D. Kispert, I. A. Nikitina, N. A. Pankrushina and A. V. Tkachev, J. Phys. Chem. B, 2010, 114, 4646–4651 CrossRef CAS PubMed.
- A. A. Schlotgauer, V. I. Klimentiev, V. S. Kornievskaya, N. E. Polyakov, A. A. Stepanov, S. F. Vasilevsky and T. V. Leshina, Appl. Magn. Reson., 2015, 46, 559–573 CrossRef CAS.
- E. C. Clark and V. Aronov, Pestic. Sci., 1996, 47, 355–362 CrossRef.
- A. Beeby and A. E. Jones, J. Photochem. Photobiol., B, 2000, 72, 10–15 CrossRef CAS.
- A. Beeby and A. E. Jones, J. Photochem. Photobiol., B, 2001, 64, 109–116 CrossRef CAS.
- R. Staudmayer and T. D. Roberts, Tetrahedron Lett., 1974, 13, 1141–1144 CrossRef.
- S. K. Jain and N. K. Jain, Int. J. Cosmet. Sci., 2010, 32, 89–98 CrossRef CAS PubMed.
- J. L. Belant, S. W. Gabrey, R. A. Dolbeer and T. W. Seamans, Crop Prot., 1995, 14, 171–175 CrossRef CAS.
- S. A. Osadchii, N. A. Pankrushina, M. M. Shakirov, E. E. Shults and G. A. Tolstikov, Russ. Chem. Bull., 2000, 3, 557–562 CrossRef.
- N. A. Pankrushina, I. A. Nikitina, N. V. Anferova, S. A. Osadchii, M. M. Shakirov, E. E. Shults and G. A. Tolstikov, Russ. Chem. Bull. Int. Ed., 2003, 52, 2490–2499 CrossRef CAS.
- A. A. Stepanov, S. F. Vasilevsky and G. A. Tolstikov, Chem. Sustainable Dev., 2010, 8, 505–510 Search PubMed.
- H. E. Ungnade, J. Am. Chem. Soc., 1954, 76, 5133–5135 CrossRef CAS.
- S. S. Ostakhov, E. M. Tsyrlina, S. G. Yunusova, M. S. Yunusov and V. P. Kazakov, Russ. Chem. Bull., 1997, 46, 705–707 CrossRef CAS.
- H. Shimada, A. Nakamura, T. Yoshihara and S. Tobita, Photochem. Photobiol. Sci., 2005, 4, 367–375 CAS.
- B. Tashkhodzhaev, Chem. Nat. Compd., 2010, 46, 421–425 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and Owner Societies 2018 |