
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visible-light-driven two-way photoisomerization of 1-(1-pyrenyl)-2-(2-quinolyl)ethylene in neutral and protonated forms†
Mikhail F.
Budyka
 * and
Vitalii M.
Li
* and
Vitalii M.
Li
Institute of Problems of Chemical Physics, Russian Academy of Sciences, pr. Akademika Semenova 1, Chernogolovka, Moscow region 142432, Russian Federation. E-mail: budyka@icp.ac.ru
First published on 8th December 2017
Abstract
Diarylethylenes with large π-systems often lose their photochemical activity (the size effect). 1-(1-Pyrenyl)-2-(2-quinolyl)ethylene (1P2QE), despite having a large conjugated π-system of 28 electrons, undergoes two-way reversible trans–cis photoisomerization both in the neutral and protonated forms with quantum yields as high as 0.13–0.83. For the neutral 1P2QE, experimental data and quantum-chemical calculations indicate a diabatic (nonadiabatic) reaction mechanism. Due to high photoisomerization quantum yields and the long-wavelength absorption band at 340–460 nm and 390–560 nm for the neutral and protonated compounds, respectively, 1P2QE can be used as a molecular photoswitch that is sensitive to visible light.
1. Introduction
Photoisomerization is one of the fundamental reactions in photochemistry. Photoisomerization of retinal is a primary photochemical step in the signal transduction cascade of vision.1,2 Photoisomerization of stilbene and azobenzene derivatives, which are the most widespread photochromes, is a functioning mechanism of many photonic molecular switches and logic gates.3–6 For example, by photoisomerization of different diarylethylenes (DAEs), it is possible to perform the functions of different molecular logic gates.7,8A molecular photonic switch is defined as a compound that can reversibly transform between two (or more) states under light irradiation. There are two main parameters that characterize the photochemical properties of a photonic switch: the region of spectral sensitivity and the photoisomerization quantum yield. Spectral sensitivity is defined by the absorption spectrum of a photochrome that is used as a photonic switch. Commonly, photoswitches, being rather small molecules, absorb light in the UV region (at least, one of the photochrome isomers); however, for many applications, the visible region is more suitable, since it reduces the damage from UV irradiation of the material.9 Therefore, the possibility of switching the photochrome using visible light has attracted increasing attention.10–13
There are several mechanisms of spectral tuning. In nature, the spectral sensitivity of the retinal photochrome is tuned over a broad wavelength range by environmental variation since the retinal absorption spectrum depends on interaction with the opsin protein, to which retinal is bound (on the polarity of the retinal binding pocket of rhodopsin, on proximity of the counter ion).1,2 Owing to this dependence, the same retinal photochrome provides color vision across the visible spectrum.
In azobenzene-based photoswitches, the spectrum is tuned by introducing electron-donating and/or electron-withdrawing substituents, which can significantly affect the π–π* and n–π* absorption bands (and simultaneously, the thermal stability).9
The most general mechanism of spectral tuning, which is used for designing visible-light photoswitches, is a decrease in the photochrome HOMO–LUMO (highest occupied – lowest unoccupied molecular orbital) gap that is achieved by extending the conjugated π-system of the photochrome.9 A smaller HOMO–LUMO gap produces a red-shifted absorption spectrum. However, photochrome, with a large π-system, often loses photochemical activity that prevents its exploitation as a photoswitch despite possessing the necessary absorption spectrum.
For example, on going from stilbene to 2-styrylnaphthalene (SN), the DAE π-system increases from 14 to 18 π-electrons and the trans → cis photoisomerization quantum yield (ϕtc) drops from ∼0.5 to 0.12.14–16 The further increase in the π-system by 4 electrons gives rise to 2-styrylanthracene (SA, 22 electrons), at which the ϕtc value reduces practically to zero.17,18 Since the cis-isomer remains reactive (the cis → trans photoisomerization quantum yield ϕct>0), the photochemical properties of SA are characterized as a ‘one-way’ cis → trans photoisomerization reaction. The sharp ϕtc decrease when the π-system exceeds some threshold size, which is the so-called ‘size effect’, is explained by the change in the reaction mechanism from diabatic (nonadiabatic) to adiabatic: upon excitation of the cis-isomer, the system ‘slides’ along the excited-state potential energy surface (PES) to the trans-isomer (adiabatic mechanism) passing the perp-conformer (phantom state), where it could branch between trans- and cis-isomers (diabatic mechanism, see below). When the trans-isomer is excited, the energy is localized on the large aromatic nucleus, which increases the barrier on the excited state PES and prevents twisting to the perp-conformer (and further to the cis-isomer).19 One-way adiabatic cis–trans photoisomerization (in the singlet-excited state) is also characteristic for 1-styrylpyrene (SP), which has 24 π-electrons.20
According to the functioning mechanism, azobenzene derivatives and DAEs are E/Z (trans/cis) photoswitches. In contrast to azobenzene derivatives, DAEs are rarely used as photoswitches despite their advantages, for example, they have thermally stable isomers separated by a large barrier in the ground (S0) state that results in fully photo-driven switching.
X. Guo et al.11 have noted that surprisingly little effort has been devoted to developing visible-light-triggered “non-azo” E/Z photoswitches. They have designed bis(dithienyl)dicyanoethene (4TCE) with absorption bands in the visible region, which is capable of isomerizing under visible light.11,21 An interesting property of 4TCE is that E/Z photoswitching operates by multiplicity-exclusive pathways: trans-to-cis isomerization only occurs in the S1 state, whereas cis-to-trans isomerization occurs in the T1 state. Moreover, the photoisomerization quantum yields vary with excitation wavelength; for example, ϕct is negligible at 530 nm and increases to 0.05 at 420 nm.21
Earlier, we have studied the size effect in a series of aza-DAEs – derivatives of 2-styrylquinoline (SQ). Upon stepwise addition of the benzene rings to the styryl fragment, we obtained 1-(1-naphthyl)-2-(2-quinolyl)ethylene 1N2QE,22 1-(2-naphthyl)-2-(2-quinolyl)ethylene 2N2QE22 and 1-(9-anthryl)-2-(2-quinolyl)ethylene 9A2QE23 (Scheme 1, only s-trans and s-cis conformers relative to the vinyl-quinoline single bond are shown; depending on the structure of the aryl group, the corresponding s-trans and s-cis conformers relative to the vinyl-aryl single bond can also exist). In this series, the π-system size increases from 18 π-electrons (SQ) to 26 π-electrons (9A2QE). In accordance with this, the long-wavelength absorption band (LWAB) in the row of SQ–2N2QE–9A2QE is bathochromically shifted in the sequence of 340–348–393 nm for the trans-isomers and 272–327–390 nm for the cis-isomers. In the same row, the photoisomerization quantum yield changes as 0.27–0.39–0.33 for the ϕtc and 0.38–0.54–0.40 for the ϕct, i.e., all these compounds are photoactive and undergo two-way photoisomerization. Therefore, in this series of aza-DAEs, the threshold size of the π-system is more than 26 electrons.
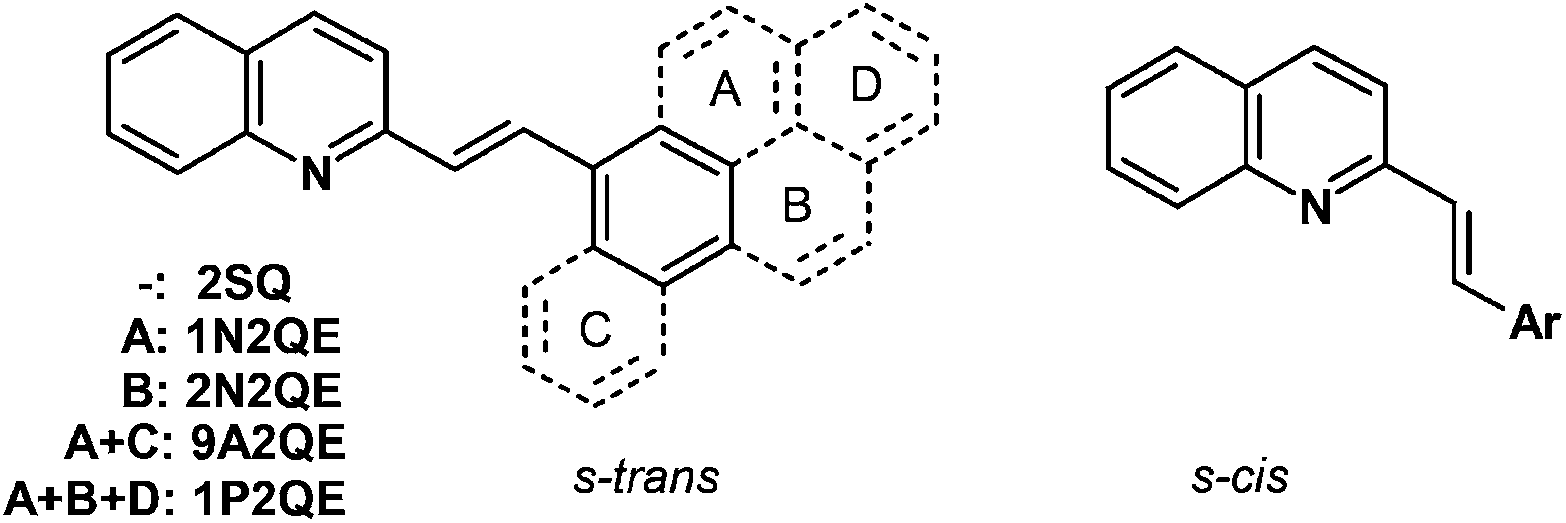 | ||
| Scheme 1 Structure of 2-styrylquinoline and its annelated derivatives (s-trans and s-cis conformers relative to the vinyl-quinoline single bond are shown). | ||
The next compound in this series where the size effect could manifest itself is 1-(1-pyrenyl)-2-(2-quinolyl)ethylene (1P2QE), which has 28 π-electrons, Scheme 1. This compound was previously synthesized,24 however, its photochemical properties have not been studied. The lack of the size effect observed previously makes the pyrene-substituted system a good candidate for pushing the spectral response into the visible region.
An additional advantage of aza-DAEs in comparison to all-carbon analogues is that they have an extra channel for spectral tuning: under protonation, their absorption spectra are red-shifted, widening the spectral sensitivity region.22,23 The effect of the nitrogen heteroatom on the photochemical properties of 1,2-diarylethenes has been previously studied.25
Considering the above discussion, in the present paper, we studied the photochemical properties of 1P2QE. In contrast to SP, 1P2QE proved to be photoactive and underwent two-way photoisomerization in the neutral and protonated forms with quantum yields of 0.13–0.83 under visible light irradiation.
2. Experimental section
1P2QE was prepared from quinaldine and 1-pyrenecarboxaldehyde by slightly modifying the previously described microwave-assisted solvent-free technique26 (see details in the ESI†).All studies were carried out in air-saturated solutions in ethanol at room temperature. The protonated form was obtained in situ by adding the required amount of HCl. Quartz cells with an optical path length of l = 1 cm were used. Electronic absorption spectra were recorded on a Specord M-400 spectrophotometer; emission spectra were recorded on a PerkinElmer LS-55 spectrofluorimeter. Fluorescence quantum yields were measured using a dilute alcohol solution of anthracene as a standard (with a fluorescence quantum yield of 0.3 (ref. 27)), with a measurement accuracy of 15%. Fluorescence lifetimes were measured by a PicoQuant GmbH instrument, using the single photon counting method.
A light-emitting diode LED-260 (emitting wavelength 260 nm, full width at half maximum FWHM = 12 nm) and a DRSh-500 high-pressure mercury lamp were used as sources of UV and visible light. The spectral lines at 313, 365, 405, 436 and 546 nm were isolated with a set of glass filters from Elektrosteklo. Light intensity of (1–5) × 10−9 Einstein cm−2 s−1 was measured by a ferrioxalate actinometer. Photoisomerization quantum yields were calculated by numerical solution of a set of differential equations and averaged in several experiments; error did not exceed 20%. All experiments were performed in a darkroom under red light.
2.1 Quantum-chemical calculations
Geometry optimizations were performed using the Gaussian 09 program package.28 The structures of the compounds in the ground state were calculated with full optimization of the geometrical parameters using semiempirical PM3 and DFT B3LYP/6-31G* methods. The optimized structures corresponded to the minima at the potential energy surfaces and had no imaginary frequencies in the vibrational spectra. The absorption spectra were calculated by the time-dependent (TD) B3LYP/6-31G* method using ground-state geometry optimized at the B3LYP/6-31G* level. In the S1 state, optimization of the geometrical parameters was performed by TD B3LYP/6-31G* and PM3 with configuration interaction PM3-CI(2 × 2). Minimal energy paths on the PESs (reaction profiles) were calculated by the PM3 (the S0 state) and PM3-CI(2 × 2) methods (the S1 state).3. Results and discussion
Fig. 1 shows the UV/Vis absorption spectra of 1P2QE in the neutral and protonated forms; the spectra of the trans-isomers are experimental, while the spectra of the cis-isomers are calculated by Fischer's method.29 The LWAB of the neutral trans-isomer has a maximum at 387 nm (ε = 3.92 × 104 L mol−1 cm−1). For the cis-isomer, a hypsochromic shift of the LWAB (characteristic for the DAEs) up to 364 nm is observed and accompanied by a decrease in intensity (ε = 2.36 × 104 L mol−1 cm−1) in comparison with the trans-isomer.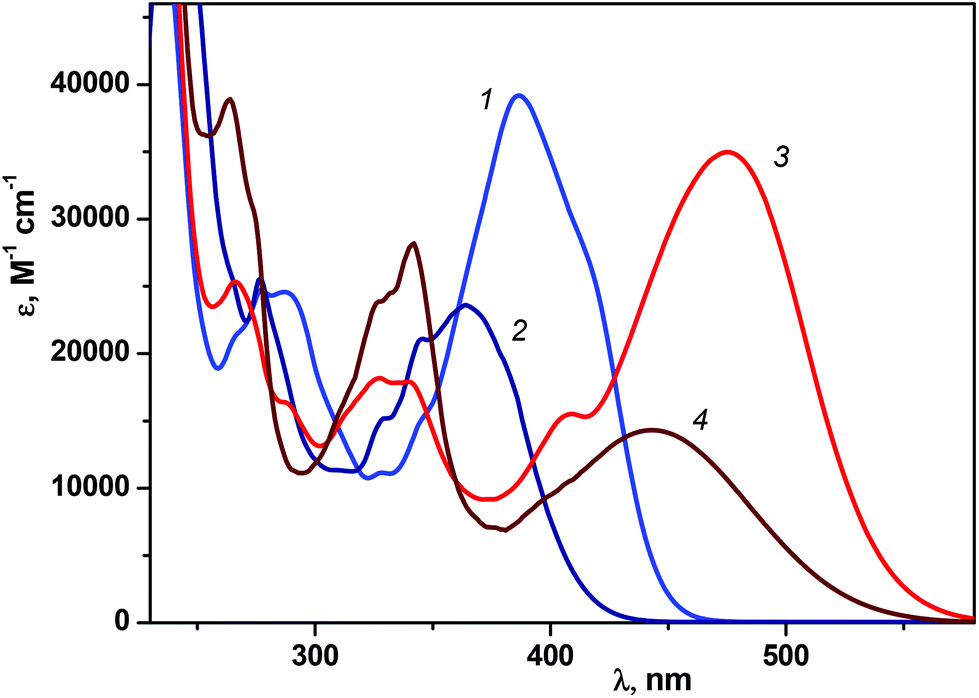 | ||
| Fig. 1 Absorption spectra (in ethanol) of 1P2QE: neutral (1, 2) and protonated (3, 4) forms, trans-isomers (1, 3) and cis-isomers (2, 4). | ||
It should be noted that pyrene itself has a very characteristic absorption spectrum with a well-defined vibrational structure (Fig. S3 in the ESI†).30 Due to the planar structure and participation of the pyrene nucleus in π-conjugation, in E-1P2QE (as well as in E-SP20), the local pyrene (S2 ← S0) transition in the region of 320–340 nm is practically masked by the S1 ← S0 transition of the 1P2QE molecule as a whole.
Since the cis-isomer has a twisted nonplanar conformation, where the conjugation between the pyrene and quinoline nuclei through the ethylene group is partially broken, the local pyrene transitions in the cis-isomer are more pronounced than in the trans-isomer, hence the spectrum of the cis-isomer is more structured with additional maxima at 346 nm and 330 nm (Fig. 1, spectrum 2).
On comparing the spectra of the neutral and protonated species (Fig. 1) the position of the local pyrene absorption bands in the region of 330–350 nm is practically independent of the charge of the quinoline nucleus (0 or +1), while for the LWAB, characterizing the entire conjugated π-system, a significant bathochromic shift in the cationic form is observed to the region of 390–560 nm with maxima at 475 nm for the trans-isomer and 444 nm for the cis-isomer, (Fig. 1, spectra 3 and 4).
The neutral E-1P2QE fluoresces with a quantum yield of φfl = 0.28 and maximum at 476 nm, the fluorescence lifetime is 1.33 ns (Fig. S4†). On protonation, a bathofloric shift of the emission band to 591 nm and a decrease in intensity by more than an order of magnitude (φfl = 0.013) are observed.
Like many DAEs, 1P2QE can exist as s-cis or s-trans conformers (rotamers) relative to the single bond between the ethylenic group and quinoline nucleus.31 The small dependence of the E-1P2QE fluorescence spectrum on the excitation wavelength (Fig. S5†) indicates the presence of different conformers in the solution. The conformers have slightly different spectral properties, while mono-exponential fluorescence decay implies that the conformers appear to have the same lifetime.
According to B3LYP/6-31G* calculations, in the ground S0 state, the E-1P2QE s-trans conformer is higher in energy than the s-cis conformer by 0.98 kcal mol−1 (in vacuum); in ethanol (polarizable continuum model, PCM), the difference decreases to 0.67 kcal mol−1. B3LYP/6-31G* predicts that the pyrene nucleus in the s-cis conformer is slightly twisted from the molecule plane (by ∼22°), while PM3 predicts a planar structure (Fig. S6†). The cis-isomer is higher in energy than the trans-isomer by 6.30 kcal mol−1 for the s-cis conformer and 4.19 kcal mol−1 for the s-trans conformer (B3LYP/6-31G* data).
The frontier molecular orbitals, HOMO and LUMO, are delocalized along the entire conjugated molecule for both the s-trans- and s-cis conformers of 1P2QE. As an example, Fig. 2 shows the structure of the frontier MOs for the s-cis conformer; the same MOs for the s-trans conformer are shown in Fig. S7.† We can observe that for both s-cis conformers, HOMO is bonding with respect to the central ethylenic bond, while LUMO is antibonding with respect to this bond (Fig. 2 and Fig. S7†). A predominant localization of HOMO, particularly for the cis-isomers, can also be noted at the pyrene nucleus, where the HOMO structure corresponds to that of unsubstituted pyrene. According to the TDDFT calculations at the B3LYP/6-31G* level, the LWAB corresponds to the S1 ← S0 excitation with exclusively (98%) HOMO → LUMO transition for both the s-trans- and s-cis conformers of 1P2QE (Table S1†). The calculated electronic absorption spectrum reproduces the experimental spectrum though the intensity of the S1 ← S0 transition is overestimated (Fig. S8†).
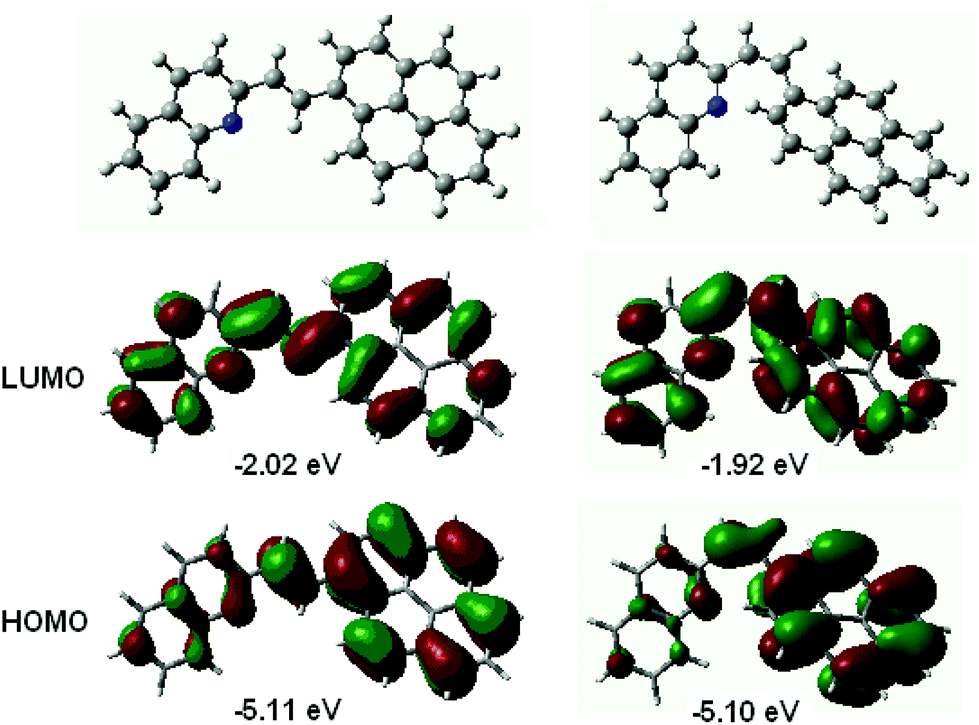 | ||
| Fig. 2 Structure and energy of the frontier molecular orbitals for the s-cis conformer of E-1P2QE (left) and Z-1P2QE (right) calculated at the B3LYP/6-31G* level. | ||
In the relaxed S1 state, according to both semiempirical and DFT methods, the E-1P2QE adopts a planar structure (Fig. S9†). The single bonds between the ethylenic group and the aromatic nuclei shorten by ∼0.03 Å, while the ethylenic double bond elongates by the same amount. This is a consequence of the electron transition from the bonding (with respect to the ethylenic bond) HOMO to the antibonding LUMO (Fig. 2 and Fig. S7†). Depopulation of the bonding MO and occupation of the antibonding MO weakens the ethylenic bond and is a prerequisite to π-bond rupture and rotation of the molecule fragments around the new single bond that can give rise to isomerization. Thus, results of the quantum-chemical calculations – the same MO structure and the same nature of the absorption band – predict the same photochemical properties for both s-conformers.
Upon irradiation of the E-1P2QE with visible light, we observed a fast reaction with spectral changes that corresponded to those for the trans–cis photoisomerization: decrease in the intensity and blue shift of the LWAB (Fig. 3). Observation of several isosbestic points at 218.4, 235.4, 279.2, 317.7 and 360.6 nm corresponded to the fact that only one reaction, namely, reversible photoisomerization, took place (Scheme 2). Principal component analysis gave a straight line with all points lying between E-1P2QE and Z-1P2QE (Fig. S10†), which corroborated the presence of only these two isomers in solution (though every isomer can exist as an equilibrated mixture of several s-conformers with a constant concentration ratio).
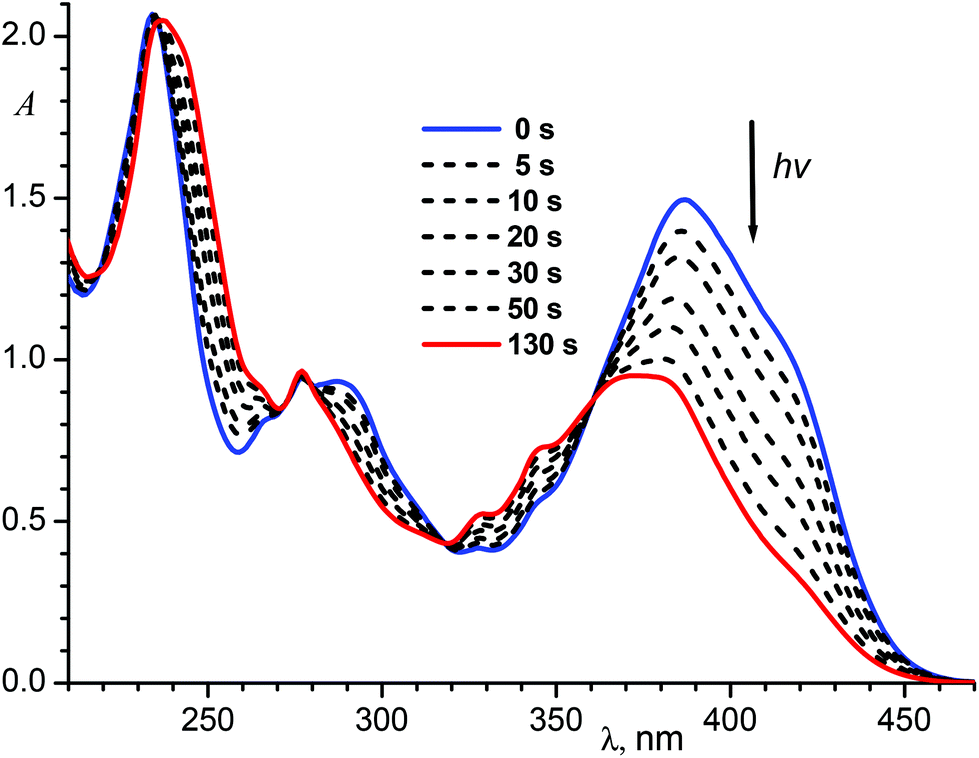 | ||
| Fig. 3 Spectral variations during irradiation of an air-saturated solution of E-1P2QE (3.8 × 10−5 M) in ethanol with 405 nm light, intensity 4.1 × 10−9 Einstein cm−2 s−1; the final spectrum corresponds to the photostationary state PS405. | ||
 | ||
| Scheme 2 Reversible photoisomerization of 1P2QE (on the example of the s-trans conformer). | ||
The spectral changes ceased on achieving the photostationary state (PS). The PSλ composition depends on the irradiation wavelength λ and is defined through the relative content of the cis-isomer:
| ηλ = [cis]PSλ/([trans]PSλ + [cis]PSλ), | (1) |
Investigation of the luminescent properties of the PSs enabled us to obtain information about the photoisomerization reaction mechanism. For the earlier studied SP, it is known that the cis-isomer possesses fluorescence and partly emits as the trans-isomer, which is adiabatically formed from the cis-isomer.20 To examine the emitting properties of the 1P2QE cis-isomer, we investigated the luminescence spectra of different PSs as well as their luminescence excitation spectra. In all cases, the PSs spectra corresponded to the trans-isomer spectra (see an example in Fig. 4).
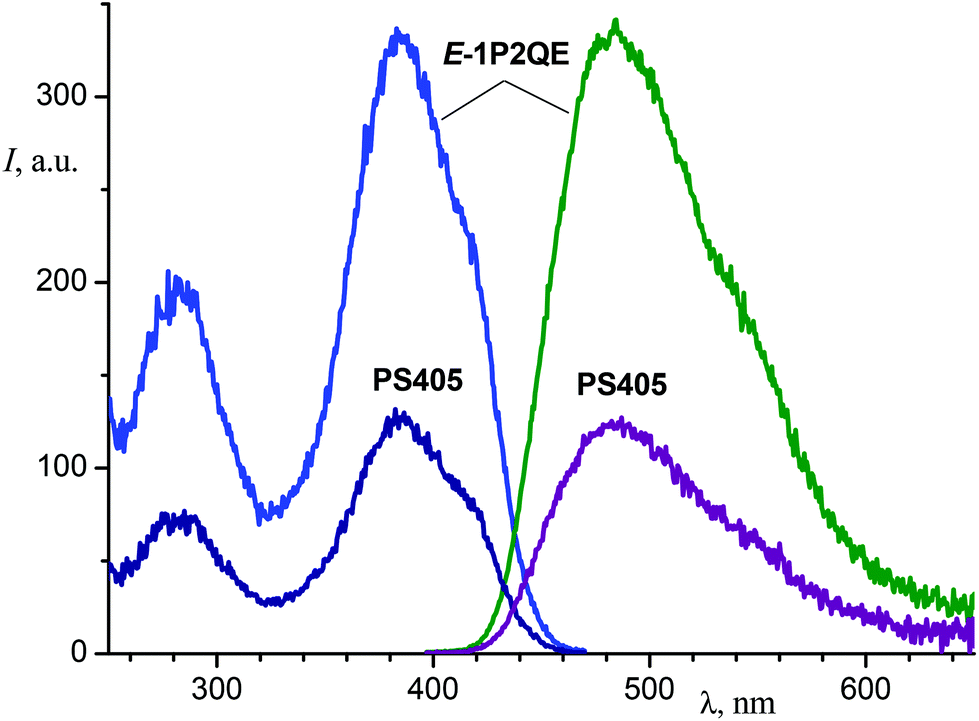 | ||
| Fig. 4 Luminescence (right, excitation at 387 nm) and luminescence excitation (left, observation at 480 nm) spectra of E-1P2QE and the photostationary state PS405 (ethanol). | ||
The luminescence intensity decreased linearly with increasing proportion of the cis-isomer in the PS (Fig. 5). The dependence was fitted by a linear regression, I/I0 = (1.002 ± 0.007)–(1.027 ± 0.015)η, with correlation coefficient R = −0.9996. Therefore, the PS fluorescence is explained exclusively by the trans-isomer emission. The excited cis-isomer does not emit luminescence either by itself or through the excited trans-isomer that could be produced as a result of adiabatic isomerization of the former. The nonradiative processes in the excited Z-1P2QE (including the chemical reaction) prevail over the radiative deactivation.
 | ||
| Fig. 5 Relative luminescence intensity of photostationary states is dependent on the PS composition (relative quantity of the cis-isomer η, see text). | ||
Absence of the adiabatically formed trans-isomer means that the photoisomerization follows diabatic mechanism that implies existence of a minimum on the excited-state PES at the geometry of the perp-conformer, see below.
Calculations of the PES cross-sections reveal that the isomerization reaction profiles coincide with the conclusion based on the experimental data. Minimal energy paths of the rotation around the ethylenic double bond (dihedral angle β) in the ground (S0) and lowest singlet-excited (S1) states for the s-cis conformer are shown in Fig. 6; for the s-trans conformer, a similar picture was obtained (Fig. S11†).
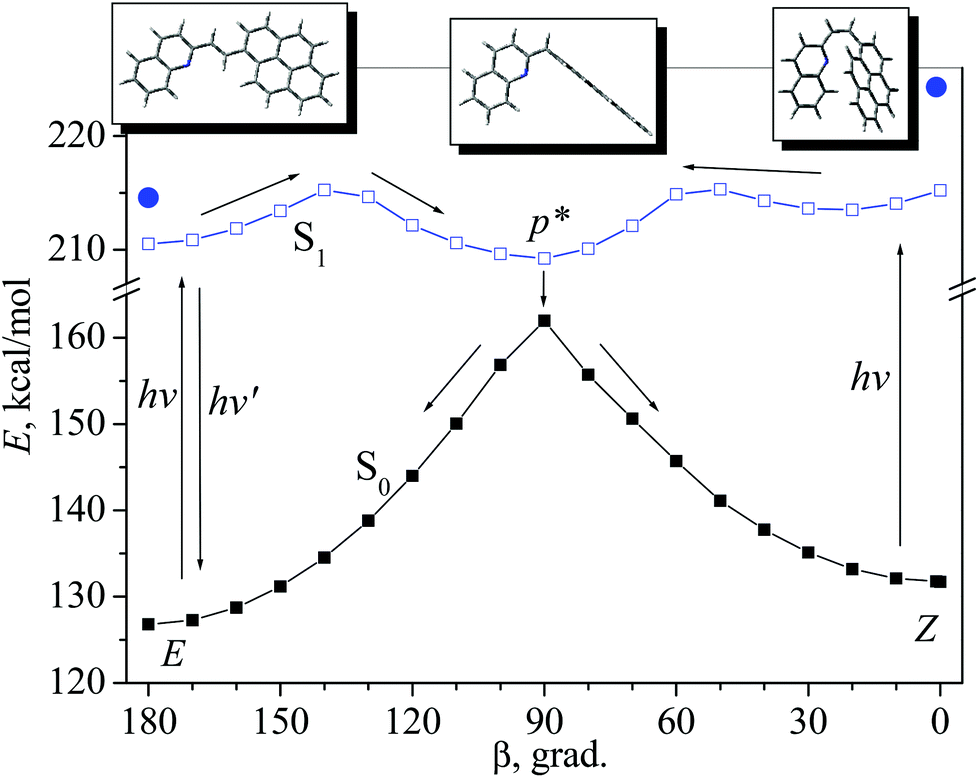 | ||
| Fig. 6 Minimal energy paths of the isomerization reaction of the 1P2QE s-cis-conformer in the S0 state (calculated by the PM3 method) and in the S1 state (calculated by the PM3-CI(2 × 2) method); the arrows show the diabatic reaction path. The filled circles mark the location of the terms of the vertically excited isomers. | ||
In the ground state, two isomers, trans-(E) and cis-(Z), are separated by a high barrier at the perp-conformer geometry (β = 90°). The existence of the high ground-state barrier explains the absence of the thermal isomerization (at room temperature). On the excited-state PES, the perp-conformer corresponds to the minimum p* (Fig. 6). After excitation, both trans- and cis-isomers achieve this minimum, and then, as a result of internal conversion (via conical intersection), the system relaxes to the ground-state maximum, where it can branch, with equal probability, between the trans- and cis-isomer; thus, the accepted value for the partitioning factor α is 0.5.19,32,33 This corresponds to the diabatic mechanism of photoisomerization.
As mentioned above, protonation of the quinoline nitrogen atom results in the LWAB bathochromic shift that allows us to use visible light with wavelength up to 560 nm for the switching between different states of the molecule.
Fig. 7 shows spectral variations under exposure of the protonated E-1P2QE to the visible light. We observed a fast photoisomerization reaction similar to the neutral compound. Isosbestic points at 235.5, 282.1, 306.0 and 358.3 nm testified about the absence of side reactions. Spectral changes also ceased on achieving the PS; using two PSs (PS436 and PS546) and the trans-isomer spectrum, the cis-isomer spectrum was calculated by Fischer's method (Fig. 1, spectrum 4).
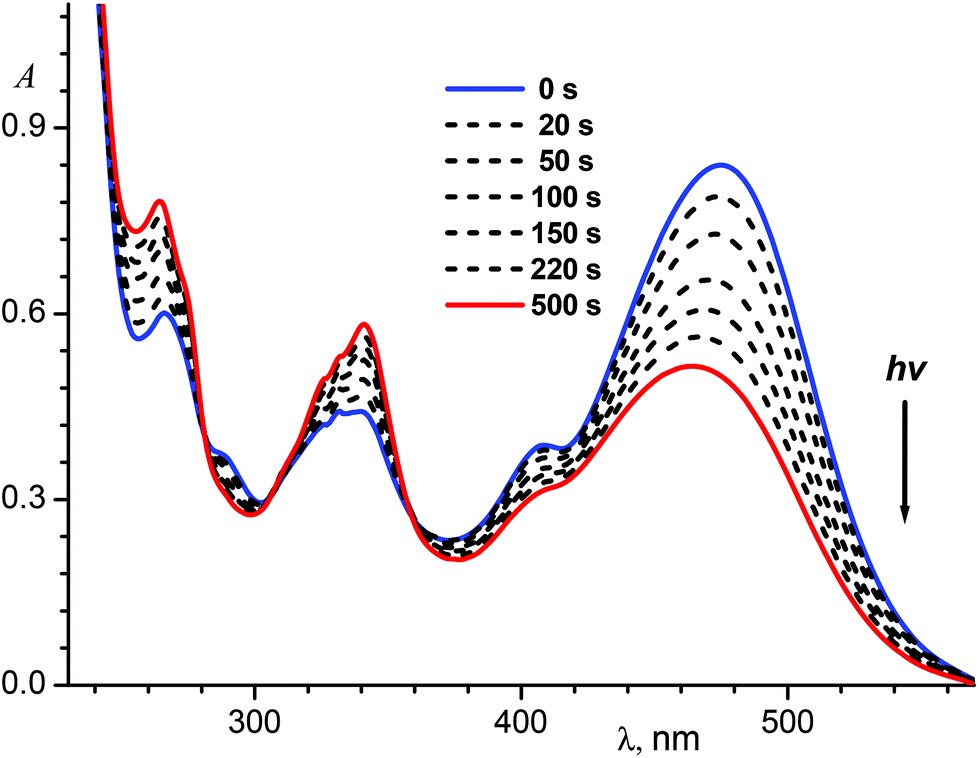 | ||
| Fig. 7 Spectral variations during irradiation of an air-saturated solution of the hydrochloride E-1P2QE·HCl (2.5 × 10−5 M) in ethanol with 546 nm light; the final spectrum corresponds to the photostationary state PS546. | ||
The change in the kinetics of absorbance (A) for a reversible trans–cis photoisomerization is described by the differential eqn (2)
| dA/dt = −(εt − εc)·(ϕtc·At − ϕct·Ac)·(1–10−A)·I0/A, | (2) |
The data obtained allow for the comparison of different pathways in the excited 1P2QE. The lifetime (τ) of the excited E-1P2QE (1.33 ns) is defined by the sum of the rate constants of three possible processes: fluorescence (kfl), nonradiative decay (knr) and twisting to the perp-conformer (ktp), τ = 1/(kfl + knr + ktp). From φfl = τkfl, we obtain kfl = 2.1 × 108 s−1. For diabatic photoisomerization, ϕtc = ατktp and therefore, ktp = 3.5 × 108 s−1 and knr = 1.9 × 108 s−1 (this can include internal conversion and intersystem crossing). Therefore, in the excited E-1P2QE, emission and nonradiative decay proceed with close efficiency, but chemical reaction is a main pathway. For Z-1P2QE, since φct/α = 1.04 (provided α = 0.5, the theoretical limiting value is 1), chemical reaction is the only pathway for this isomer within an error of the quantum yield measurement. This explains the absence of the cis-isomer fluorescence.
The data for E-1P2QE can be compared with those for E-SP, which does not photoisomerize in the trans → cis direction (ϕtc ∼0). The lifetime for the E-SP is 5.3 ns (hexane) and φfl = 0.82;35 thus, kfl = 1.5 × 108 s−1 and knr = 0.4 × 108 s−1.
Owing to effective photoisomerization, both neutral and cationic forms of 1P2QE can be used as molecular switches that are sensitive to visible light. However, it should be noted that for a molecular switch based on DAE E/Z isomerization, complete switching from one state to another is impossible because reversibility of the photoisomerization reaction and overlap of the isomer spectra prevent 100% conversion of one isomer to another upon exposure to light. As noted above, on irradiation with light of wavelength λ, a photostationary state PSλ is achieved, whose composition is defined by a photostationary equilibrium condition (3).
| (φtc·εt·[trans]PS)λ = (φct·εc·[cis]PS)λ | (3) |
According to condition (3), the lower the quantum yield, the greater is the concentration of the respective isomer in the PS. From eqn (3), we can obtain the dependence of the cis-isomer content in the PS on the irradiation wavelength (4).
| ηλ = (φtc·εt/(φtc·εt + φct·εc))λ | (4) |
The variation of ηλ with λ enables us to estimate a range of possible switching abilities of any DAE as a molecular photoswitch.
The calculated curves for the neutral and protonated 1P2QE are shown in Fig. 8. For 1P2QE, the quantum yield ratio does not depend on the irradiation wavelength, so the dependence of ηλ on λ is defined exclusively by the MAC ratio εt/εc. The maximum ηλ dependence is observed at a wavelength where the εt/εc ratio achieves a maximal value; conversely, the minimum on the ηλ dependence corresponds to the minimal εt/εc value. Thus, for the neutral 1P2QE, the maximal (94%) and minimal (18%) content of the cis-isomer can be obtained at 441 nm and 251 nm, respectively (Fig. 8, curve 1).
 | ||
| Fig. 8 Dependence of the photostationary state composition (relative quantity of the cis-isomer ηλ, see text) on the irradiation wavelength for the neutral (1) and protonated (2) 1P2QE. | ||
A necessary property of a molecular photoswitch for application in optoelectronic devices is photostability with respect to side reactions. 1P2QE has high fatigue resistance. For example, Fig. 9 shows the changes in absorbance upon alternating irradiation with visible and UV light. The total time of irradiation was 4400 s, every cycle corresponded to the switching between two PSs: PS365 (η365 = 33%) and PS436 (η436 = 94%).
 | ||
| Fig. 9 Changes in absorbance upon alternating irradiation of an air-saturated solution of E-1P2QE in ethanol with UV (365 nm, 200 s) and visible (436 nm, 200 s) light: 1 – at 387 nm (the E-1P2QE absorption maximum), 2 – at 436 nm. High (low) absorbance corresponds to the photostationary state PS365 (PS436). | ||
3. Conclusion
1-(1-Pyrenyl)-2-(2-quinolyl)ethylene (1P2QE) has a rather large conjugated π-system of 28 electrons. Nevertheless, it undergoes two-way reversible trans → cis and cis → trans photoisomerization with quantum yields as high as ϕtc = 0.23 and ϕct = 0.52 for the neutral 1P2QE, and ϕtc = 0.13 and ϕct = 0.83 for the protonated 1P2QE. For the neutral 1P2QE, both experimental data and quantum-chemical calculations testify the diabatic mechanism of photoisomerization. The neutral 1P2QE has a long-wavelength absorption band in the region of 340–460 nm with a maximum at 387 nm for the trans-isomer and 364 nm for the cis-isomer. Upon protonation, the absorption spectrum is red-shifted to 390–560 nm with a maximum at 475 nm for the trans-isomer and 444 nm for the cis-isomer. Due to high photoisomerization quantum yields and absorption in the visible region, 1P2QE can be used as a molecular photoswitch that is sensitive to visible light. The switching ability of 1P2QE is restricted by possible changes in the photostationary composition between the ηλ values (the cis-isomer content) in the range of 18–94% for the neutral form and 9–45% for the cationic form. To avoid possible deprotonation of the cationic form in basic media, the N-alkylated derivatives can be used.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Russian Foundation for Basic Research (Grant No. 17-03-00789).References
- I. Rivalta, A. Nenov and M. Garavelli, Modelling retinal chromophores photoisomerization: from minimal models in vacuo to ultimate bidimensional spectroscopy in rhodopsins, Phys. Chem. Chem. Phys., 2014, 16, 16865–16879 RSC.
- H. Kandori, Y. Shichida and T. Yoshizawa, Photoisomerization in Rhodopsin, Biochemistry, 2001, 66, 1197–1209 CAS.
- K. Szacilowski, Digital Information Processing in Molecular Systems, Chem. Rev., 2008, 108, 3481–3548 CrossRef CAS PubMed.
- M. F. Budyka, Molecular Photonic Logic Gates, High Energy Chem., 2010, 44, 121–126 CrossRef CAS.
- U. Pischel, J. Andreasson, D. Gust and V. F. Pais, Information Processing with Molecules - Quo Vadis?, ChemPhysChem, 2013, 14, 28–46 CrossRef CAS PubMed.
- M. F. Budyka, Molecular switches and logic gates for information processing, the bottom-up strategy: from silicon to carbon, from molecules to supermolecules, Russ. Chem. Rev., 2017, 86, 181–210 CrossRef CAS.
- M. F. Budyka, N. I. Potashova, T. N. Gavrishova and V. M. Li, Design of Fully Photonic Molecular Logic Gates Based on the Supramolecular Bisstyrylquinoline Dyad, Nanotechnol. Russ., 2012, 7, 280–287 CrossRef.
- M. F. Budyka and V. M. Li, Multifunctional Photonic Molecular Logic Gate Based On A Biphotochromic Dyad With Reduced Energy Transfer, ChemPhysChem, 2017, 18, 260–264 CrossRef CAS PubMed.
- D. Bleger and S. Hecht, Visible-Light-Activated Molecular Switches, Angew. Chem., Int. Ed., 2015, 54, 11338–11349 CrossRef CAS PubMed.
- S. Helmy, F. A. Leibfarth, S. Oh, J. E. Poelma, C. J. Hawker and J. Read de Alaniz, Photoswitching Using Visible Light: A New Class of Organic Photochromic Molecules, J. Am. Chem. Soc., 2014, 136, 8169–8172 CrossRef CAS PubMed.
- X. Guo, J. Zhou, M. A. Siegler, A. E. Bragg and H. E. Katz, Visible-Light-Triggered Molecular Photoswitch Based on Reversible E/Z Isomerization of a 1,2-Dicyanoethene Derivative, Angew. Chem., Int. Ed., 2015, 54, 4782–4786 CrossRef CAS PubMed.
- S. Fredrich, R. Gostl, Ma. Herder, L. Grubert and S. Hecht, Switching Diarylethenes Reliably in Both Directions with Visible Light, Angew. Chem., Int. Ed., 2016, 55, 1208–1212 CrossRef CAS PubMed.
- T. van Leeuwen, J. Pol, D. Roke, S. J. Wezenberg and B. L. Feringa, Visible-Light Excitation of a Molecular Motor with an Extended Aromatic Core, Org. Lett., 2017, 19, 1402–1405 CrossRef CAS PubMed.
- U. Mazzucato, Photophysical and photochemical behaviour of stilbene-like molecules and their aza-analogues, Pure Appl. Chem., 1982, 54, 1705–1721 CrossRef CAS.
- S. Malkin and E. Fischer, Temperature Dependence of Photoisomerization. III. Direct and Sensitized Photoisomerization of Stilbenes, J. Phys. Chem., 1964, 68, 1153–1163 CrossRef CAS.
- J. Saltiel, A. Marinari, D. W.-L. Chang, J. C. Mitchener and E. D. Megarity, Trans-Cis Photoisomerization of the Stilbenes and a Reexamination of the Positional Dependence of the Heavy-Atom Effect, J. Am. Chem. Soc., 1979, 101, 2982–2996 CrossRef CAS.
- A. Spalletti and G. Bartocci, Temperature and solvent effects on rotamer-specific photobehaviour of the cis and trans isomers of 2-styrylanthracene, Phys. Chem. Chem. Phys., 1999, 1, 5623–5632 RSC.
- J. Saltiel, Y. Zhang and D. F. Sears Jr., Temperature Dependence of the Photoisomerization of cis-1-(2-Anthryl)-2-phenylethene. Conformer-Specificity, Torsional Energetics and Mechanism, J. Am. Chem. Soc., 1997, 119, 11202–11210 CrossRef CAS.
- M. F. Budyka, Diarylethene photoisometization and photocyclization mechanisms, Russ. Chem. Rev., 2012, 81, 477–493 CrossRef CAS.
- A. Spalletti, G. Bartocci, U. Mazzucato and G. Galiazzo, Decay pathways of the first excited singlet state of cis-1-styrylpyrene, Chem. Phys. Lett., 1991, 186, 297–302 CrossRef CAS.
- J. Zhou, X. Guo, H. E. Katz and A. E. Bragg, Molecular Switching via Multiplicity-Exclusive E/Z Photoisomerization Pathways, J. Am. Chem. Soc., 2015, 137, 10841–10850 CrossRef CAS PubMed.
- M. F. Budyka, N. I. Potashova, O. V. Chashchikhin, T. N. Gavrishova and V. M. Li, Photoisomerization of Naphthylquinolylethylenes in Neutral and Protonated Forms, High Energy Chem., 2011, 45, 492–496 CrossRef CAS.
- M. F. Budyka, N. I. Potashova, T. N. Gavrishova and V. M. Li, Photochemical Properties of 1-(9-Anthryl)-2-(2-Quinolyl)ethylene, High Energy Chem., 2014, 48, 185–190 CrossRef CAS.
- G. Drefahl, K. Ponsold and E. Gerlach, Untersuchungen über Stilbene, XXVII. Stilbazole mit höheren aromatischen Ringsystemen, Chem. Ber., 1960, 93, 481–485 CrossRef CAS.
- S. Ciorba, F. Fontana, G. Ciancaleoni, T. Caronna, U. Mazzucato and A. Spalletti, Fluorescence/photoisomerization competition in trans-aza-1,2-diarylethenes, J. Fluoresc., 2009, 19, 759–768 CrossRef CAS PubMed.
- V. M. Li, T. N. Gavrishova and M. F. Budyka, Microwave-Assisted Solvent-Free Synthesis of 2-Styrylquinolines in the Presence of Zinc Chloride, Russ. J. Org. Chem., 2012, 48, 823–828 CrossRef CAS.
- H. D. Becker, Unimolecular photochemistry of anthracenes, Chem. Rev., 1993, 93, 145–172 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- E. Fischer, The Calculation of Photostationary States in Systems A<=>B When Only A Is Known, J. Phys. Chem., 1967, 71, 3704–3706 CrossRef CAS.
- J. Duhamel, Internal Dynamics of Dendritic Molecules Probed by Pyrene Excimer Formation, Polymers, 2012, 4, 211–239 CrossRef CAS.
- U. Mazzucato and F. Momicchioli, Rotational Isomerism in trans-1,2-Diarylethylenes, Chem. Rev., 1991, 91, 1679–1719 CrossRef CAS.
- V. D. Vachev, J. H. Frederick, B. A. Grishanin, V. N. Zadkov and N. I. Koroteev, Quasiclassical Molecular Dynamics Simulation of the Photoisomerization of Stilbene, J. Phys. Chem., 1995, 99, 5247–5263 CrossRef CAS.
- G. Bartocci, U. Mazzucato and A. Spalletti, The role of the adiabatic mechanism in the photoisomerization of 1,2-diarylethenes and related compounds, Trends Phys. Chem., 2007, 12, 1–36 CAS.
- M. F. Budyka, N. I. Potashova, T. N. Gavrishova and V. M. Li, Photoisomerization of 2-Styrylquinoline in Neutral and Protonated Forms, High Energy Chem., 2008, 42, 446–453 CrossRef CAS.
- Y. Kikuchi, H. Okamoto, T. Arai and K. Tokumaru, Effects of solvents and substituents controlling the adiabatic or diabatic modes of the cis ->trans isomerization of styrylpyrenes in the excited singlet state, Chem. Phys. Lett., 1994, 229, 564–570 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, experimental and calculated absorption spectra, fluorescence decay profile, dependence of fluorescence spectra on excitation wavelength, principal component analysis, transition energies, nature and oscillator strengths of the lowest excited singlet states calculated by TDDFT/B3LYP, calculated optimized geometries in the S0 and S1 states, minimal energy paths calculated by PM3 and PM3-CI (2 × 2). See DOI: 10.1039/c7pp00359e |
| This journal is © The Royal Society of Chemistry and Owner Societies 2018 |