
The effect of the functional ionic group of the viologen derivative on visible-light driven CO2 reduction to formic acid with the system consisting of water-soluble zinc porphyrin and formate dehydrogenase†
S.
Ikeyama
a and
Y.
Amao
 *ab
*ab
aThe Advanced Research Institute for Natural Science and Technology, Osaka City University, 3-3-138 Sugimoto, Sumiyoshi-ku, Osaka 558-8585, Japan. E-mail: amao@ocarina.osaka-cu.ac.jp
bResearch Centre for Artificial Photosynthesis (ReCAP), Osaka City University, 3-3-138 Sugimoto, Sumiyoshi-ku, Osaka 558-8585, Japan
First published on 14th November 2017
Abstract
The effect of the functional ionic group of 4,4′-bipyridinium salt derivatives (4,4′-BPs) as the electron carrier on the visible-light driven conversion of CO2 to formic acid with the system consisting of water-soluble zinc tetraphenylporphyrin tetrasulfonate (ZnTPPS) and formate dehydrogenase (FDH) in the presence of triethanolamine (TEOA) as an electron donor was investigated. 1,1′-Diaminoethyl- (DAV), 1-aminoethyl-1′-methyl- (AMV), 1-carboxymethyl-1′-methyl- (CMV) and 1,1′-dicarboxymethyl-4,4′-bipyridinium salt (DCV) were prepared as the 4,4′-BPs with the functional ionic group. Irradiation of a CO2 saturated buffer solution containing TEOA, ZnTPPS, 4,4′-BP and FDH with visible light irradiation resulted in the production of formic acid. By using 4,4′-BPs with the cationic aminoethyl-group, DAV or AMV as an electron carrier, the effective visible-light driven formic acid production based on the CO2 reduction was observed compared to the 4,4′-BPs with the anionic carboxymethyl-group, CMV or DCV. The formic acid production rate with DAV was approximately 3.2 times higher than that of the system with DCV.
Introduction
A photoredox system consisting of an electron donating molecule, a visible-light sensitizer, an electron carrier molecule and a catalyst is widely used for visible-light driven energy conversion systems such as hydrogen production, CO2 reduction, substrate conversion and so on.1–15 This system is a simplification of the visible-light driven electron transfer process in a natural photosynthesis reaction.16 Water soluble zinc porphyrins, ruthenium polypyridyl coordination complexes and Mg chlorophyll-a are useful visible-light sensitizers in these systems.17–20 The development of an effective catalyst is also an important key factor for an effective visible-light driven energy conversion system. Among the catalysts used for visible-light driven energy conversion systems, some biocatalysts have received much attention. For visible-light driven hydrogen production, hydrogenase (H2ase) has been used as a hydrogen producing catalyst.21–30 For visible-light driven CO2 reduction or substrate conversion, various dehydrogenases such as lactate (LDH),31–33 formate (FDH),34–36 aldehyde (AldDH)37–39 and alcohol dehydrogenase (ADH)40–42 for the production of valuable organic compounds such as lactic acid,43 formic acid44 and methanol45–47 have been used as catalysts. These biocatalysts are classified into NAD+-dependent dehydrogenases. Among these dehydrogenases, FDH catalyzes formic acid oxidation to CO2 with the co-enzyme NAD+ and catalyzes the reverse reaction of CO2 reduction to formic acid with the co-enzyme NADH. In particular, FDH from Candida boidinii is commercially available and is widely used in visible-light driven CO2 reduction. Thus, visible-light driven CO2 conversion to a formic acid system is developed with the combination of NAD+/NADH redox coupling and FDH as shown in Fig. 1.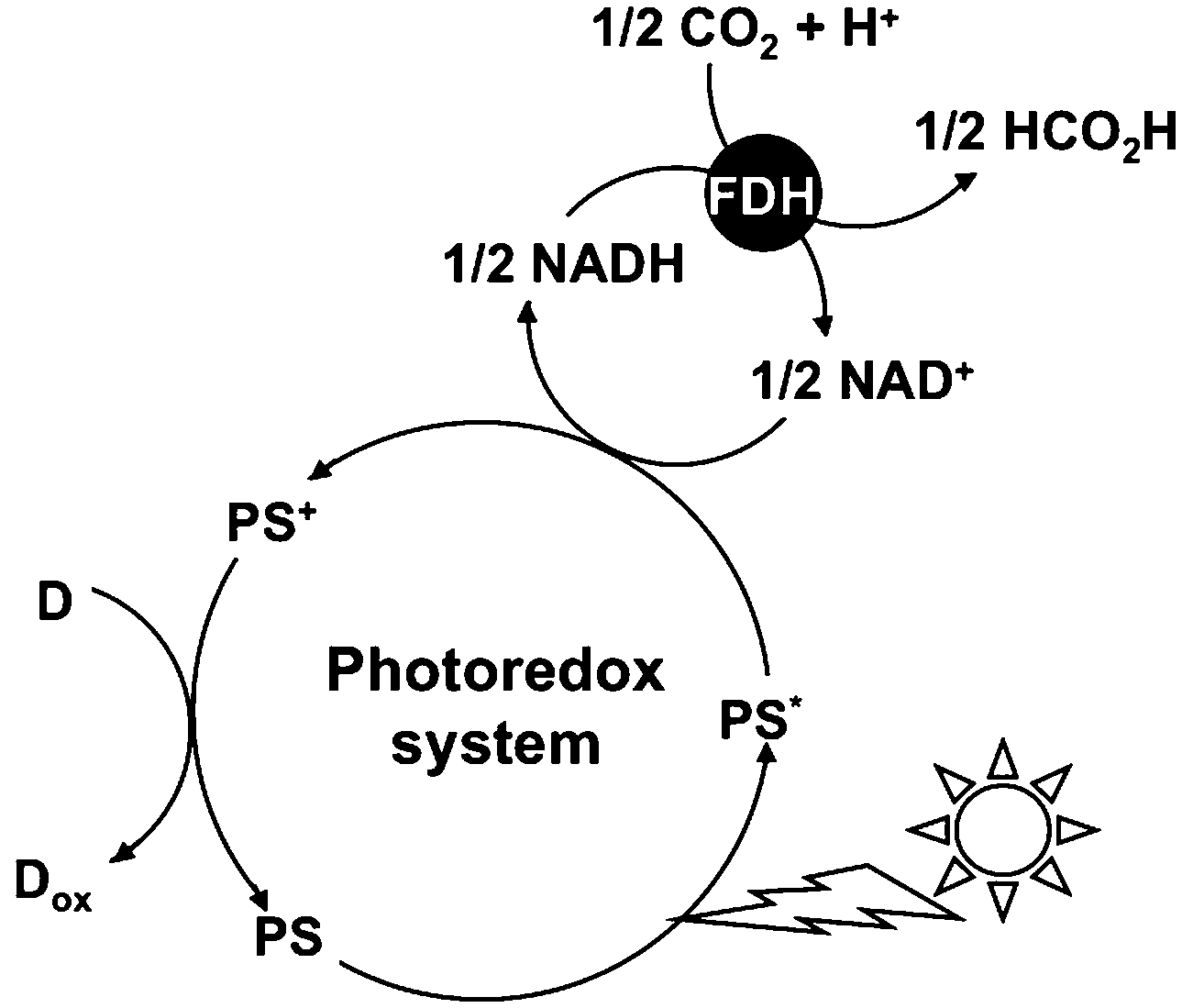 | ||
| Fig. 1 Visible light-driven CO2 reduction to formic acid with the photoredox system consisting of an electron donor (D), a photosensitizer (PS), NAD+ (an electron carrier) and FDH. | ||
However, the NAD dimer is formed between the one-electron reduced form of NAD+s with a visible-light sensitizer such as tris(bipyridine)ruthenium(II), Ru(bpy)32+.48 The NAD dimer is an inactive co-enzyme for NAD+-dependent FDH. Thus, it is quite difficult to construct the visible-light driven conversion of CO2 to formic acid using a photoredox system based on the combination of NAD+ photoreduction and FDH. In contrast, some studies on the visible-light driven CO2 reduction to formic acid with FDH using the photoreduction of NAD+ by a catalyst via electron carriers such as a rhodium complex have been reported as shown in Fig. 2.49–55
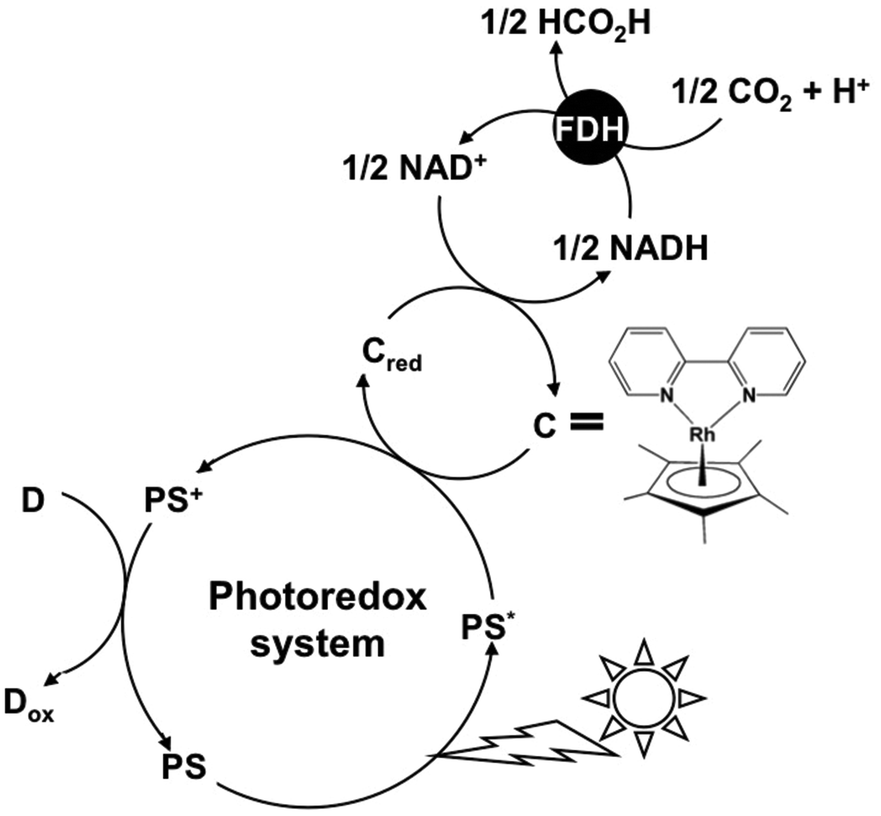 | ||
| Fig. 2 Visible light-driven CO2 reduction to formic acid with the photoredox system consisting of an electron donor (D), a photosensitizer (PS), an electron carrier, NAD+ as a second electron carrier and FDH. | ||
In this system, NAD dimerization is suppressed using the redox coupling rhodium complex. Even if the photoreduction of NAD+ to NADH could be achieved, the affinity between NADH and FDH such as Michaels-Menten constant Km, catalytic constant kcat and so on does not change, and thus, the FDH catalytic activity cannot be controlled in the photoredox system for CO2 reduction. In particular, we found that the catalytic efficiency, kcat/Km, value for the CO2 reduction to formic acid was approximately 900 times lower than that of the formic acid oxidation to CO2, catalyzed with FDH using a native co-enzyme NADH or NAD+.56 Therefore, the redox coupling of the NAD+/NADH free system is desired for the photoredox system with FDH.
In contrast, the visible-light driven CO2 reduction to formic acid systems with the coupling of the photoreduction of various 4,4′- or 2,2′-bipyridinium salts (4,4′- or 2,2′-BPs) with Ru(bpy)32+, water soluble zinc porphyrin or Mg chlorophyll-a and FDH has been reported.57–66 In this system, the single-electron reduced form of BPs acts as a co-enzyme for FDH instead of NADH. Surprisingly, the direct interaction between the single-electron reduced form of BPs and FDH has not been clarified using enzymatic kinetic analysis. Recently, we have reported the kinetic parameters of the single-electron reduced form of various 4,4′-67–70 and 2,2′-BPs56,67,71 for the CO2 reduction to formic acid with FDH using an enzymatic kinetic analysis. For the system using 2,2′-BPs, the catalytic activity of FDH for CO2 reduction is dependent on the dihedral angles between the pyridine rings in the single-electron reduced form of 2,2′-BPs.44,67,71 For the system using 4,4′-BPs with the ionic functional group (chemical structures are shown in Fig. 3), in contrast, the effective CO2 reduction to formic acid with FDH using 1,1′-diaminoethyl-4,4′-bipyridinium dichloride (DAV) was accomplished, compared with the other 4,4′-BPs.67–70
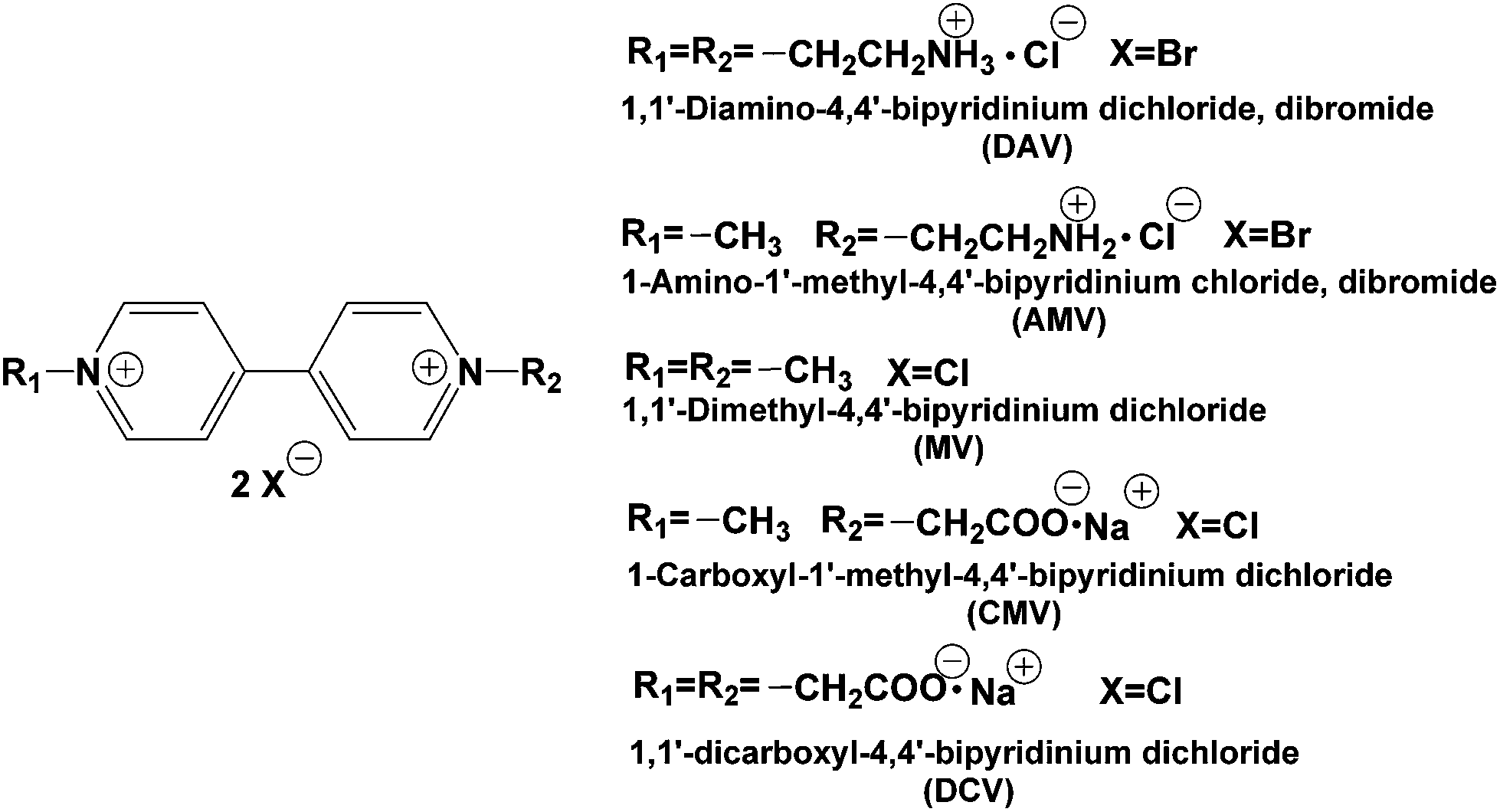 | ||
| Fig. 3 Chemical structures of various 4,4′-BPs. | ||
The catalytic efficiency of FDH with the single-electron reduced form of DAV was estimated to be more than 560 times higher than that of the NADH.67,69,70 As a communication, we previously reported that the DAV is an effective electron carrier for the visible light-driven photoredox system for formic acid production from CO2 with the system consisting of FDH and zinc tetraphenylporphyrin tetrasulfonate (ZnTPPS) in the presence of TEOA as shown in Fig. 4.72
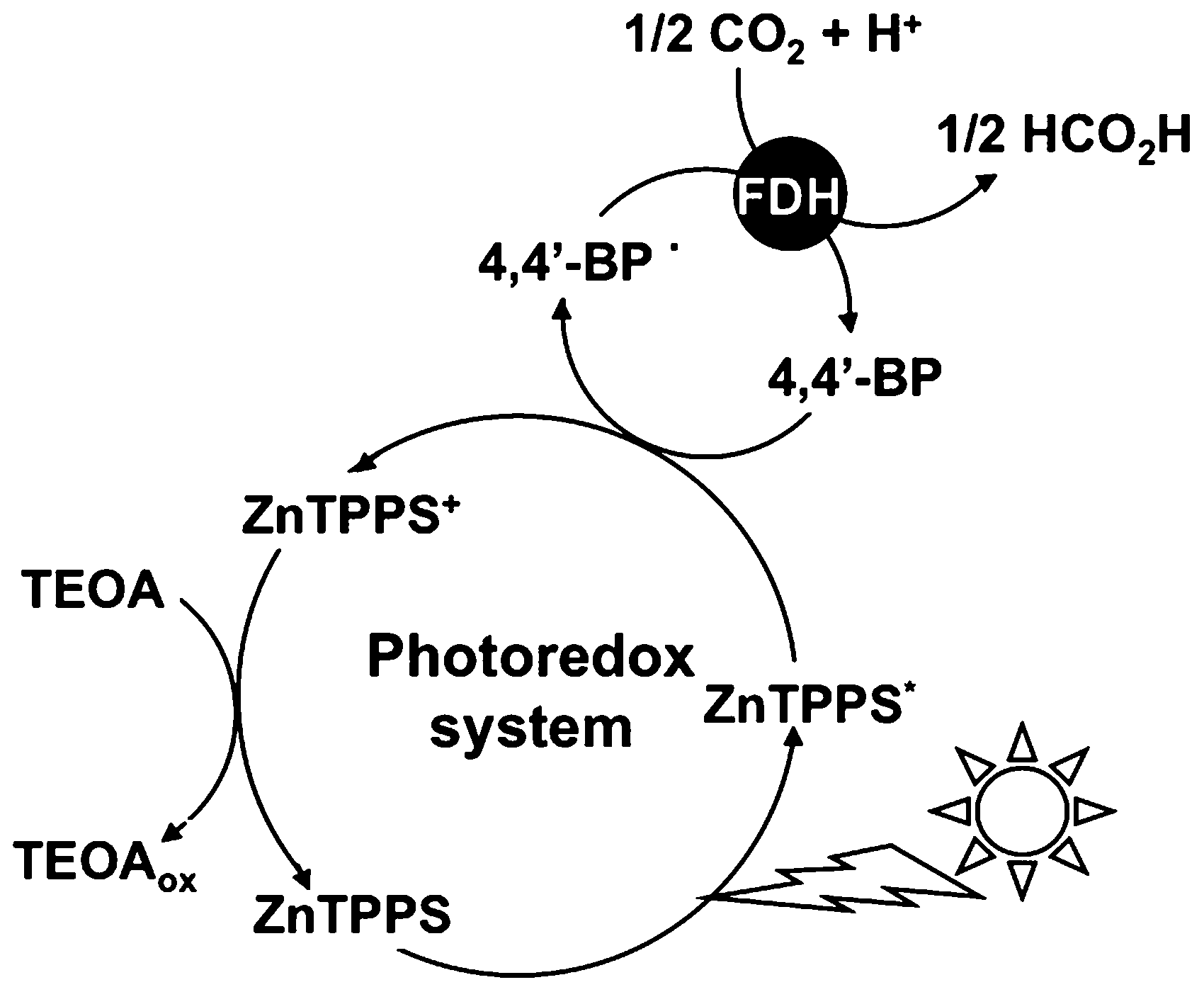 | ||
| Fig. 4 Visible light-driven CO2 reduction to formic acid with the photoredox system consisting of TEOA, ZnTPPS, 4,4′-BP and FDH. | ||
In this paper, more detailed effects of various 4,4′-BPs with cationic or anionic functional groups on visible-light driven CO2 reduction to formic acid with the system consisting of ZnTPPS and FDH in the presence of TEOA were studied.
Experimental
Materials
FDH from Candida boidinii was purchased from Roche Diagnostics K.K. The molecular weight of FDH from Candida boidinii was estimated to be 74 kDa.73 MV2+ dichloride, 4,4′-bipyridine, 1-bromoacetic acid and 2-bromoethylamine hydrobromide were purchased from Tokyo Kasei Co. Ltd. The other chemical reagents were of analytical grade or the highest grade available. Tetraphenylporphyrin tetrasulfonate (H2TPPS), TEOA and methyl iodide were supplied by Tokyo Kasei Co. Ltd. The other chemicals were analytical grade or the highest grade available. ZnTPPS was synthesized by refluxing H2TPPS with about 10 times molar equivalent of zinc acetate according to the previously reported literature.45,46,64 As a small amount of zinc acetate was not removed from ZnTPPS solution prepared, it has been confirmed that zinc acetate has no influence on the following photoreactions.17,18 Moreover, the stability of ZnTPPS against visible-light irradiation was checked by the following experiment. The solution of ZnTPPS (100 μM) in 3.0 ml of 1.0 mM sodium pyrophosphate buffer (pH 7.4) was deaerated by freeze–pump–thaw cycles, repeated 6 times. The sample solution was irradiated with a 250 W halogen lamp (TOSHIBA JD110V215WNP-EH-TB) at a distance of 5.0 cm at 30 °C. The degradation of ZnTPPS was determined by the absorbance change at 422 nm due to ZnTPPS with a Shimadzu Multispec 1500 spectrophotometer. For 7 h continuous irradiation, little change in the absorbance at 422 nm was observed.64 Thus, ZnTPPS has a good stability for continuous irradiation.Preparation of 4,4′-BPs
DAV or DCV was synthesized from 4,4′-bipyridine and 1-bromoacetic acid or 2-bromoethylamine hydrobromide according to the previously reported literature.70 AMV or CMV was synthesized from 1-methyl-4,4′-bipyridinium iodide and bromoacetic acid or 2-bromoethylamine hydrobromide according to the previously reported literature.70 The purities of 4,4′-BPs prepared were checked using proton nuclear magnetic resonance spectroscopy with a 1H-NMR Varian GEMINI-200. 1H-NMR in D2O: δ (ppm) DAV: 2.99 (t, 4H), 4.64 (t, 4H), 8.32 (m, 4H), 9.02 (m, 4H); AMV: 2.99 (t, 2H), 4.30 (s, 3H), 4.64 (t, 2H), 8.30–8.36 (m, 4H), 9.00–9.05 (m, 4H); CMV: 4.30 (s, 3H), 5.39 (s, 2H), 8.30–8.36 (m, 4H), 9.00–9.05 (m, 4H); DCV: 5.39 (s, 4H), 8.33 (m, 4H), 8.96 (m, 4H). The chemical shifts were referenced to the solvent peak calibrated against tetramethylsilane (TMS).Determination of molar coefficient for single-electron reduced form of 4,4′-BPs
Molar coefficients for the single-electron reduced form of 4,4′-BPs were determined by the following method. A solution containing 4,4′-BP and sodium dithionite (0.1 M) in distilled water was deaerated with argon gas bubbling for 5 min. The production of the single-electron reduced form of 4,4′-BP was monitored by the UV-vis absorption spectrum. Molar coefficients for the single-electron reduced form of 4,4′-BP were obtained from the slope in the plot of the concentration of single-electron reduced form of 4,4′-BP and the absorbance at 605 nm in the absorption spectrum using the Beer–Lambert law.Reduction potential measurements of 4,4′-BPs
The single and double-electron reduction potentials for 4,4′-BP2+s were determined by cyclic voltammetry (Hokuto Denko HZ-3000). All measurements were carried out under a nitrogen-saturated solution containing 0.2 M potassium chloride and 1.0 mM sodium pyrophosphate buffer (pH 7.4) at a glassy carbon-working electrode. A platinum wire was used as a counter electrode. All potentials were relative to the Ag/AgCl electrode used as the reference. The final concentration of 4,4′-BPs was adjusted to 2.0 mM.Calculation for dihedral angle between the pyridine rings in the oxidized and single-electron reduced form of 4,4′-BPs
The dihedral angle between the pyridine rings in the oxidized and single-electron reduced form of 4,4′-BPs was calculated by the Molecular Mechanics Program 2 (MM2). The MM2 proposed by Allinger was the method based on classical mechanics to model molecular systems. The potential energy of all systems in molecular mechanics is calculated based on the force fields. The basic calculation method was based on the following properties. (1) Each atom was simulated as one particle, (2) each particle was assigned typically the van der Waals radius, polarizability, and a constant net charge, and (3) bonded interactions were treated as springs with an equilibrium distance equal to the experimental or calculated bond length.74Visible-light driven reduction of 4,4′-BP with photosensitization of ZnTPPS
A solution consisting of TEOA (0.3 M), ZnTPPS (10 μM) and 4,4′-BP (100 μM) in 5.0 ml of 10 mM sodium pyrophosphate buffer (pH 7.4) was deaerated by freeze–pump–thaw cycles, repeated 5 times. The sample solution was irradiated with a 250 W halogen lamp (TOSHIBA JD110V215WNP-EH-TB) at a distance of 5.0 cm at 30 °C. The production of the single-electron reduced form of 4,4′-BP was monitored by the UV-Vis absorption spectrum.Visible-light driven formic acid production from CO2 with the system consisting of ZnTPPS, 4,4′-BP and FDH
A solution consisting of TEOA (0.3 M), ZnTPPS (10 μM), 4,4′-BP (100 μM) and FDH (6.4 μM) in 5.0 ml of 10 mM sodium pyrophosphate buffer (pH 7.4) was deaerated by freeze–pump–thaw cycles repeated 5 times and the gas phase was replaced with CO2. The sample solution was irradiated with a 250 W halogen lamp at 30 °C. The amount of formic acid was detected by ion chromatography (Dionex ICS-1100; electrical conductivity detector) with an ion exclusion column (Thermo ICE AS1; column length: 9 × 150 mm; composed of 7.5 μm cross-linked styrene/divinylbenzene resin with functionalized sulfonate groups). 1.0 mM octane sulfonic acid and 5.0 mM tetrabutylammonium hydroxide were used as an eluent and a regenerant, respectively.Results and discussion
Determination of molar coefficient for single-electron reduced form of 4,4′-BPs
Molar coefficients for single-electron reduced form of 4,4′-BPs were determined by the absorbance maximum at 605 nm in absorption spectra using the Beer–Lambert law. The colour in 4,4′-BP solution changed from colorless to blue or violet with the production of the single-electron reduced form of 4,4′-BPs. From the results of absorbance at maxima in the spectra, molar coefficients for the single-electron reduced form of DAV, AMV, CMV and DCV were determined to be 4200, 4500, 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 18000 M−1 cm−1, respectively. As a reference, the molar coefficients for the single-electron reduced form of MV at 605 nm was 13000 M−1 cm−1. From these results, the carboxy- or amino-group was affected by the molar coefficients for the single-electron reduced form of 4,4′-BPs.
000 and 18000 M−1 cm−1, respectively. As a reference, the molar coefficients for the single-electron reduced form of MV at 605 nm was 13000 M−1 cm−1. From these results, the carboxy- or amino-group was affected by the molar coefficients for the single-electron reduced form of 4,4′-BPs.
Calculation of the dihedral angle between the pyridine rings in the oxidized and single-electron reduced form of 4,4′-BPs
Fig. S1† shows the structures of the oxidized and the single-electron reduced form of 4,4′-BPs. In all 4,4′-BPs, the dihedral angles between the pyridine rings in the oxidized form and the single-electron reduced form of 4,4′-BPs were estimated to be 0 and 90°, respectively. We previously reported that the dihedral angles between the pyridine rings in 2,2′-BPs were important factors for the activation of the catalytic activity of CO2 reduction of FDH. As the single-electron reduced form of DB2+ with a small dihedral angle easily interacts with the binding site of FDH due to small steric hindrance, the effective formic acid production from CO2 with FDH was attained.For 4,4′-BPs, in contrast, there is no difference in the dihedral angles between the pyridine rings of the single-electron reduced form of 4,4′-BPs.
In the case of AMV and CMV, the radical position was localized onto the methyl-group side or aminoethyl-group side for AMV, and carboxymethyl-group side for CMV. In the case of AMV, there is no difference in the radical positions in the dihedral angles between the pyridine rings of the single-electron reduced form. For the case of CMV, on the other hand, the conformational structure of the single-electron reduced form of CMV with the radical position localized onto the carboxymethyl-group side was drastically changed compared to that of the radical position localized onto the methyl-group side.
Reduction potentials for the single and double-electron reduced form of 4,4′-BPs
Table 1 shows the reduction potentials for the single and double-electron reduced form of 4,4′-BPs (vs. Ag/AgCl). We previously reported that single-electron reduction potentials for DAV, AMV, MV, CMV and DCV are estimated to be −0.60, −0.62, −0.67, −0.67 and −0.67 V (vs. Ag/AgCl), respectively.70 In contrast, the double-electron reduction potentials for DAV, AMV, MV, CMV and DCV are estimated to be −0.91, −0.94, −0.98, −0.98 and −0.99 V (vs. Ag/AgCl), respectively.| 4,4′-BPs | Single-electron reduction potential (V) | Double-electron reduction potential (V) |
|---|---|---|
| DAV | −0.60 | −0.91 |
| AMV | −0.62 | −0.94 |
| MV | −0.67 | −0.98 |
| CMV | −0.67 | −0.98 |
| DCV | −0.67 | −0.99 |
The single and double-electron reduction potentials of 4,4′-BPs were influenced by the bonded functional groups. For example, the single and double-electron reduction potentials of 1,1′-diphenyl-4,4′-bipyridinium salt (DPV) was estimated to be −0.39 and −0.74 V (vs. Ag/AgCl), respectively. The difference in the single and double-electron reduction potentials between DPV and MV was estimated to be 0.24 and 0.27 V, respectively. This result indicates that the phenyl-group directly affects the reduction potential of 4,4′-BPs by the electron-withdrawing effect. On the other hand, the difference in the single and double-electron reduction potentials of 4,4′-BPs in this study was estimated to be only about ±0.08 V. This result indicates that the bonded aminoethyl- or carboxymethyl-group to 4,4′-BPs is not affected by the single and double-electron reduction potentials of 4,4′-BPs.
Visible-light driven reduction of 4,4′-BPs with the sensitization of ZnTPPS
When the solution consisting of TEOA, ZnTPPS and 4,4′-BP was irradiated with visible-light, the absorption spectra of the solution were changed with irradiation time. Fig. 5 shows the absorption difference spectral change with the solution as a baseline before irradiation. In all 4,4′-BPs, the absorption band at 605 nm due to the single-electron reduced form of 4,4′-BPs increased with increasing irradiation time. Thus, the single-electron reduction of 4,4′-BPs proceeded with the visible-light sensitization of ZnTPPS.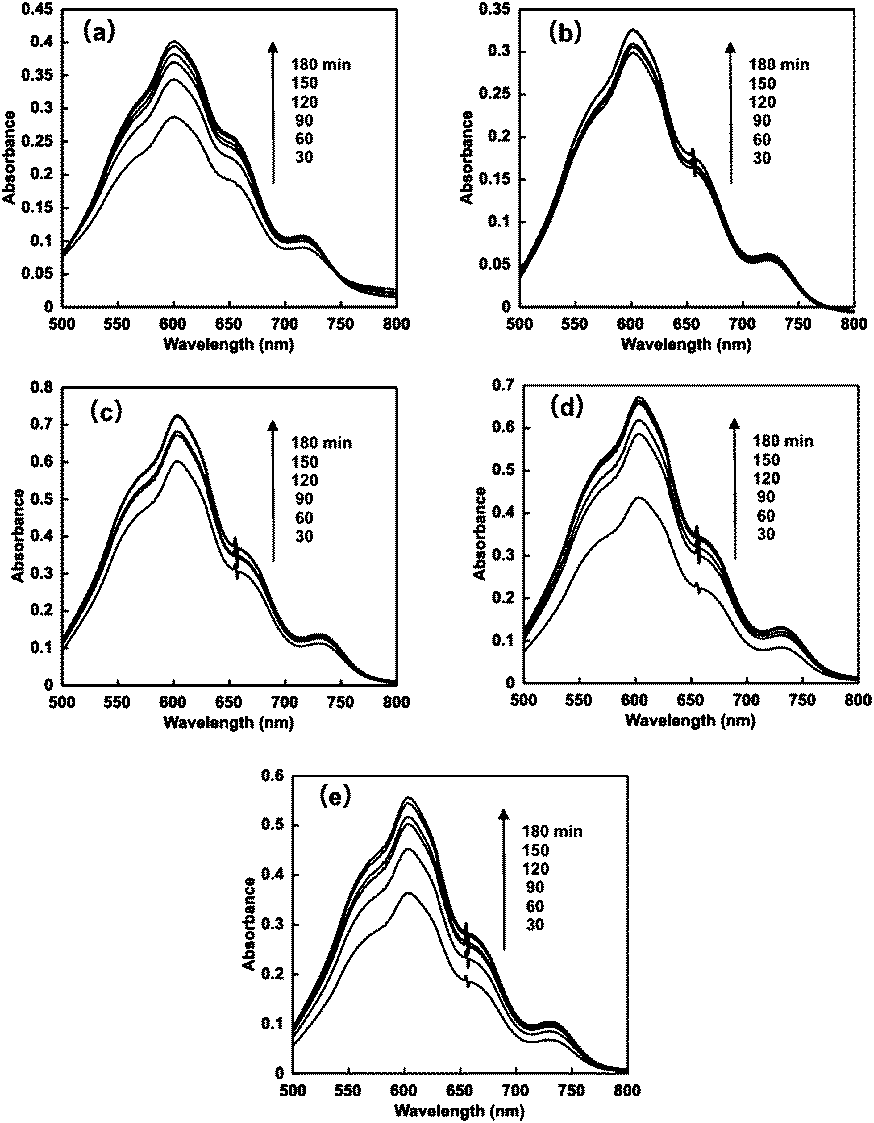 | ||
| Fig. 5 Absorption difference spectral changes in the solution consisting of TEOA, ZnTPPS and 4,4′-BPs with visible-light irradiation. (a) DAV, (b) AMV, (c) CMV, (d) DMV and (e) MV. | ||
The time dependence of the concentration of the single-electron reduced form of 4,4′-BPs produced was determined by the molar coefficient of the single-electron reduced form of 4,4′-BPs at 605 nm. Fig. 6 shows the relationship between the irradiation time and the concentration of the single-electron reduced form of 4,4′-BPs.
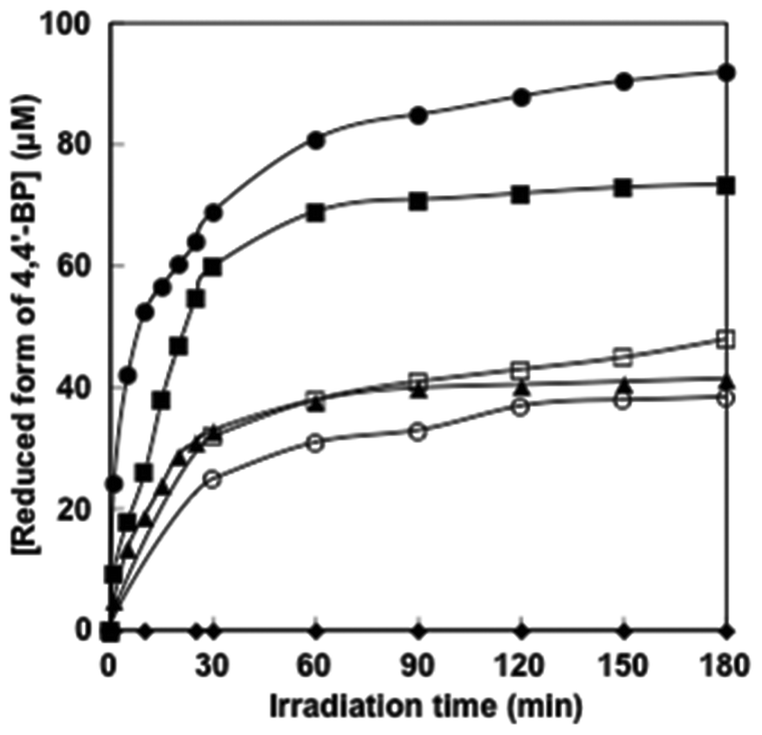 | ||
| Fig. 6 Time dependence of the single-electron reduced form of 4,4′-BP concentration in the solution consisting of TEOA, ZnTPPS and 4,4′-BPs with visible-light irradiation. (●) DAV, (■) AMV, (□) CMV, (○) DMV, (▲) MV and (◆) NAD+. | ||
In all cases, the concentration of the single-electron reduced form of 4,4′-BPs increased with increasing irradiation time. When the solution consisting of TEOA, ZnTPPS and NAD+ was irradiated with visible-light, in contrast, no photoreduction of NAD+ to NADH was observed. The initial rate was determined from the gradient of the linear part of the single-electron reduced form of 4,4′-BP production within 10 min irradiation. The initial rates for the reduced form of DAV, AMV, CMV, DCM and MV production were estimated to be 5.2, 3.9, 1.7, 1.0 and 1.9 μM min−1, respectively. After 180 min irradiation, the concentration of the single-electron reduced form of DAV, AMV, MV, CMV and DCV was estimated to be 94.5, 72.3, 46.2, 36.8 and 42.4 μM, respectively and the conversion yields of the single-electron reduced form of DAV, AMV, CMV, DCM and MV production from the oxidized form of each 4,4′-BP were estimated to be 0.945, 0.723, 0.462, 0.368 and 0.424, respectively. The DAV was almost completely reduced with the sensitization of ZnTPPS after 180 min irradiation. The rate of the single-electron reduced form of DAV production was the largest compared with other 4,4′-BPs. Moreover, in contrast, the amount of the single-electron reduced form of DCV was the lowest among the other 4,4′-BPs. The results for the visible-light driven reduction of 4,4′-BPs with the sensitization of ZnTPPS are summarized in Table 2.
| 4,4′-BPs | TON of ZnTPPS (h−1) | v 0 (μM min−1) | Total 4,4′-BP˙ production (μM) | Yield of 4,4′-BP to 4,4′-BP˙ conversion |
|---|---|---|---|---|
| DAV | 3.15 | 5.2 | 94.5 | 0.945 |
| AMV | 2.41 | 3.9 | 72.3 | 0.723 |
| CMV | 1.54 | 1.7 | 46.2 | 0.462 |
| DCV | 1.23 | 1.0 | 36.8 | 0.368 |
| MV | 1.45 | 1.9 | 43.4 | 0.434 |
Kaneko and co-workers reported the electron transfer process from the photoexcited state of ZnTPPS to MV.75 For the steady state fluorescence spectrum measurement, the fluorescence intensity of ZnTPPS decreased with increasing MV concentrations. Thus, the fluorescence of ZnTPPS was quenched by MV. For the fluorescence lifetime measurement, in contrast, no change in the fluorescence decay curve of ZnTPPS was observed in the presence of MV. It was concluded that the electron transfer proceeded by the static mechanism due to the electrostatic effect between ZnTPPS and MV. Moreover, we reported the mechanism of the electron transfer process from the excited triplet state of ZnTPPS (3ZnTPPS*) to MV using laser flash photolysis.76
In the present system, the visible-light driven electron transfer from 3ZnTPPS* to 4,4′-BP also proceeded. The oxidation potential for TEOA was reported to be 0.93 V (vs. Ag/AgCl).77 The energy level of the first singlet state of ZnTPPS was estimated to be 2.07 eV using the average value of the frequencies of the longest wavelength of the absorption maxima and the shortest wavelength of the fluorescence emission maxima (606 nm by exciting ZnTPPS at 422 nm). The redox potentials of 3ZnTPPS*, E (ZnTPPS+/3ZnTPPS*), was reported to be −0.75 V (vs. Ag/AgCl).78,79 The single-electron reduction potentials for DAV, AMV, CMV, DCM and MV by the cyclic voltammetry measurement (vs. Ag/AgCl as a reference) were estimated to be −0.60, −0.62, −0.67, −0.67 and −0.67 V, respectively. The energy diagram for the visible-light driven electron transfer from 3ZnTPPS* to 4,4′-BP is shown in Fig. 7.
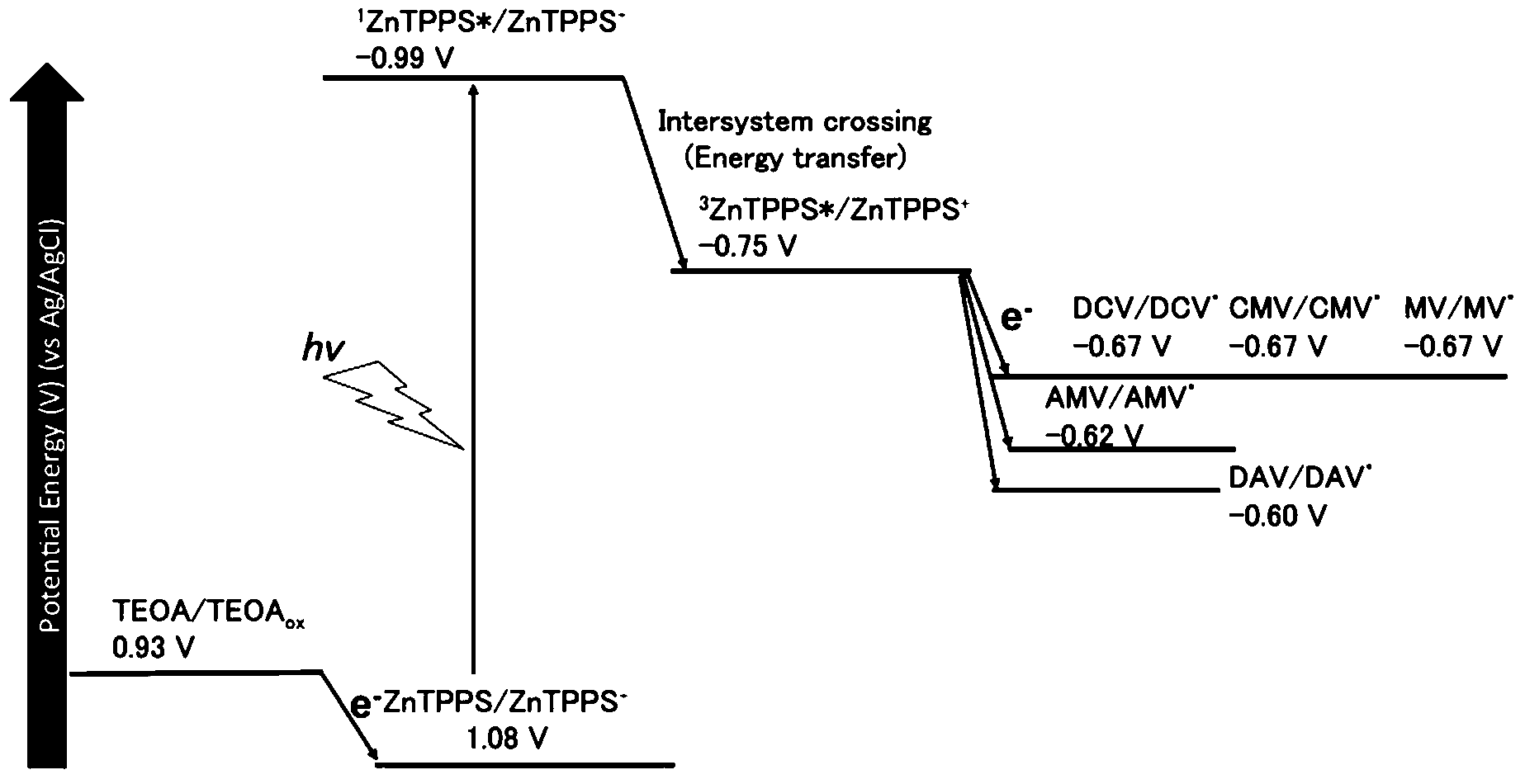 | ||
| Fig. 7 The energy diagram for the visible-light driven electron transfer from 3ZnTPPS* to 4,4′-BPs. | ||
Gibbs free energy (ΔG) for visible-light driven electron transfer from 3ZnTPPS* to 4,4′-BP was calculated using the redox potentials of 3ZnTPPS* and 4,4′-BPs. The ΔG values for visible-light driven electron transfer from 3ZnTPPS* to DAV, AMV, CMV, DCM and MV were estimated to be −14.3, −12.6, −7.72, −7.72 and −7.72 kJ mol−1, respectively. From the ΔG values, the DAV and AMV were easily reduced by 3ZnTPPS*compared with the other 4,4′-BPs.
As there is little difference among 4,4′-BPs in the single-electron reduction potentials, the photoreduction yield of 4,4′-BP to 4,4′-BP˙ conversion after 180 min irradiation increased with increasing the number of positive-electron charge of 4,4′-BP, indicating that the electron charge of BP affects the reduction yield of 4,4′-BP with photosensitization of anionic ZnTPPS. Therefore, the yield of photoreduction of 4,4′-BP was discussed from the viewpoint of intermolecular interaction between ZnTPPS and 4,4′-BP at the ground state. The intermolecular interaction between ZnTPPS and 4,4′-BP was calculated using the MM2 method. For the calculation of the interaction between ZnTPPS and 4,4′-BP, the counter ions of each molecule were ignored. Fig. 8 shows the intermolecular interaction between ZnTPPS and 4,4′-BPs at the ground state calculated by the MM2 method.
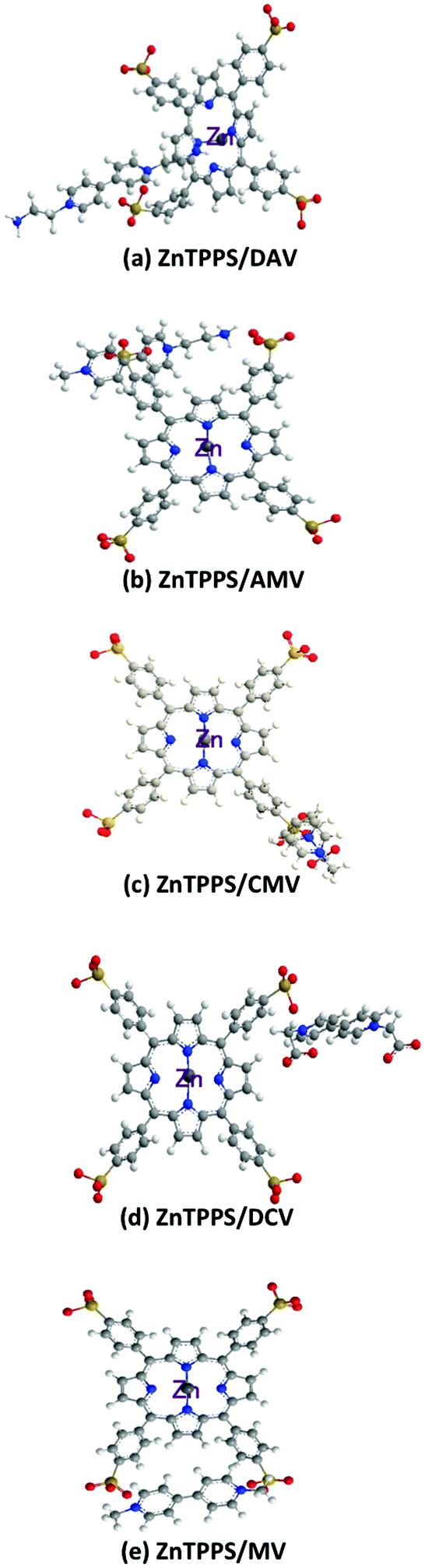 | ||
| Fig. 8 Proposed conformation based on the intermolecular interaction between ZnTPPS and 4,4′-BPs (a: DAV, b: AMV, c: CMV, d: DCV and e: MV) at the ground state calculated by the MM2 method. | ||
For the intermolecular interaction between ZnTPPS and MV, two positively charged pyridinium moieties in MV interacted with two neighbouring sulfonate-groups in ZnTPPS by the electrostatic effect. Thus, the MV was located outside the porphyrin ring and the central zinc ion in ZnTPPS at the ground state.
For the intermolecular interaction between ZnTPPS and AMV, CMV or DCV, positively charged pyridinium moieties in 4,4′-BPs interacted with a neighbouring sulfonate-group in ZnTPPS by the electrostatic effect. Thus, AMV, CMV or DCV was also located outside the porphyrin ring and the central zinc ion in ZnTPPS at the ground state.
For the intermolecular interaction between ZnTPPS and DAV, on the other hand, one of the positively charged pyridinium moieties in DAV interacted with the sulfonate-group in ZnTPPS by the electrostatic effect. The amino-group of DAV also interacted with the central zinc ion in ZnTPPS as shown in Fig. 8(a). Thus, the DAV was strongly interacted to ZnTPPS in the ground state, compared with those of the other 4,4′-BPs. Although these results were speculative, they suggested that the structural conformation based on the intermolecular interaction between ZnTPPS and DAV was the suitable structure for the visible-light driven electron transfer from ZnTPPS to DAV, compared with that of the conformation based on the interaction between ZnTPPS and AMV, CMV, DCV, or MV. The factor of improving the reduction yield of 4,4′-BP with photosensitization of ZnTPPS is the suppression of the back electron transfer process from the single-electron reduced form of 4,4′-BP to the single-electron oxidized form of ZnTPPS. From the interaction between ZnTPPS and 4,4′-BP, the formation of strong ion-pair between ZnTPPS and 4,4′-BP was induced by the suppression of the charge recombination based on the back electron transfer process from the single-electron reduced form of 4,4′-BP to the single-electron oxidized form of ZnTPPS. From Fig. 8, there was also the formation of a strong ion-pair between ZnTPPS and DAV compared with the other 4,4′-BPs. Thus, the yield of visible-light driven reduction of DAV with ZnTPPS increased compared with those of the other 4,4′-BPs. Now, we are currently planning experiments using the transition absorption spectrum measurement with laser flash photolysis for the photoinduced electron transfer process between ZnTPPS and 4,4′-BP.
Visible-light driven CO2 reduction to formic acid with the system consisting of ZnTPPS, 4,4′-BPs and FDH
Visible-light driven formic acid production from CO2 was investigated by adding FDH to a 4,4′-BP photoreduction system with photosensitization of ZnTPPS in the presence of TEOA. When the reaction mixture consisting of ZnTPPS, 4,4′-BP, TEOA and FDH in CO2 saturated sodium pyrophosphate buffer was irradiated with visible-light at 30 °C formic acid production was observed with irradiation time as shown in Fig. 9.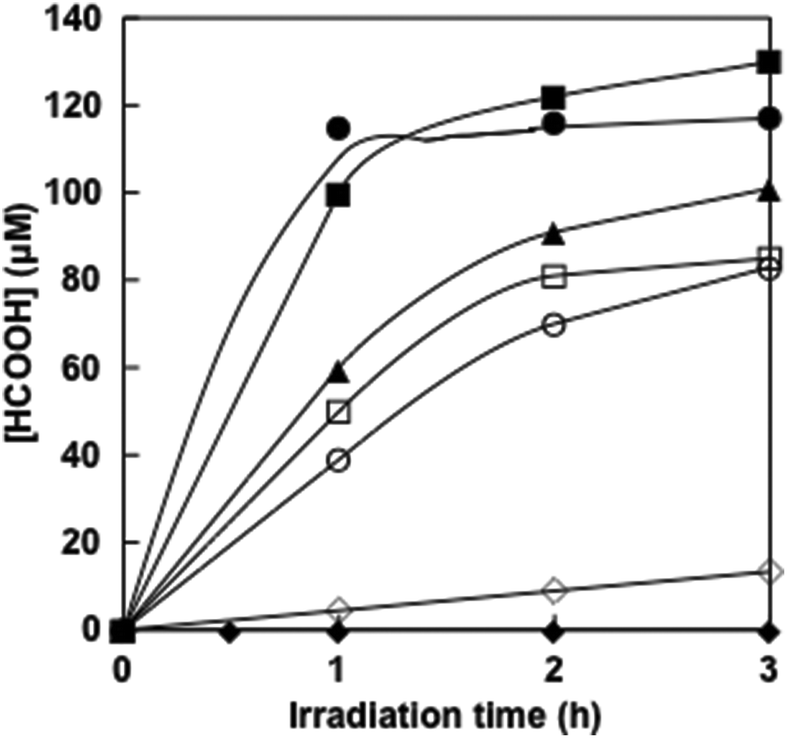 | ||
| Fig. 9 Time dependence of formic acid concentration in the solution consisting of TEOA, ZnTPPS, 4,4′-BPs and FDH with visible-light irradiation. (●) DAV, (■) AMV, (□) CMV, (○) DMV, (▲) MV and (◆) NAD+. (◊): the system consisting of NADH (100 μM) and FDH (6.4 μM) in CO2 saturated sodium pyrophosphate buffer in the dark. | ||
The concentrations of formic acid production after 3 h irradiation with DAV, AMV, CMV, DCV and MV were 120 ± 3.5, 130 ± 5.0, 85 ± 1.0, 83 ± 0.9 and 101 ± 1.5 μM, respectively. The experimental error was calculated as an average of three experiments. In all experiments, the error was within 5% and the reproducibility was satisfactory. In contrast, no formic acid production was observed in the dark or in the absence of CO2 conditions. The effects of the above four components, TEOA, ZnTPPS, 4,4′-BP and FDH on the CO2 reduction to formic acid were investigated. It was found that formic acid was not produced even if one of the above five components was missing, namely, TEOA, ZnTPPS, 4,4′-BP, FDH and CO2.
When the solution consisting of TEOA, ZnTPPS, NAD+ and FDH in CO2 saturated buffer solution was irradiated with visible-light, in contrast, no formic acid was produced with the increase in irradiation time. As no NADH was produced with the visible-light sensitization of ZnTPPS, CO2 reduction with FDH did not proceed. Moreover, the time dependence of formic acid production with the system consisting of NADH (100 μM) and FDH (6.4 μM) in CO2 saturated sodium pyrophosphate buffer under dark conditions is also shown in Fig. 9 (◊). After 3 h of incubation, 13.3 μM of formic acid was produced using natural co-enzyme NADH and FDH in the dark. The formic acid production efficiency with the visible-light driven system via the reduction of 4,4′-BPs was dramatically higher than the dark reaction with NADH.
Next, we discuss the comparison of the formic acid production rate in the visible-light driven system via the reduction of 4,4′-BPs. The rate was determined from the concentration of formic acid production within 1 h irradiation. The rates for the formic acid production with DAV, AMV, CMV, DCM and MV were estimated to be 120, 100, 50, 39 and 60 μM h−1, respectively.
For the system consisting of TEOA, ZnTPPS and FDH with DAV, AMV, CMV, DCV or MV, the amount of formic acid production was estimated to be 0.60, 0.52, 0.26, 0.20 and 0.31 μmol after 1 h irradiation. For example using DAV, it indicated that 1.2 μmol of protons and 0.6 μmol of CO2, that was 24.0 times the amount of ZnTPPS (50 nmol) in the sample solution, were reduced to formic acid. The turnover numbers of ZnTPPS for the system using AMV, CMV, DCV and MV were estimated to be 20.8, 10.4, 8.0 and 12.0 h−1, respectively. Therefore, ZnTPPS appeared to serve as a system for transferring electrons from TEOA to a more reductive molecule in all cases using 4,4′-BPs. Moreover, the turnover numbers of DAV, AMV, CMV, DCV and MV were estimated to be 1.2, 1.0, 0.52, 0.4 and 0.6 h−1, respectively. The turnover numbers of FDH in the system using DAV, AMV, CMV, DCV and MV were estimated to be 18.8, 16.3, 8.14, 6.26 and 9.45 h−1, respectively. The turnover numbers of ZnTPPS, 4,4′-BPs and FDH, and total formic acid production after 3 h irradiation with visible light-driven CO2 reduction to formic acid with FDH and ZnTPPS are summarized in Table 3.
| 4,4′-BPs | TON of ZnTPPS (h−1) | TON of 4,4′-BPs (h−1) | TON of FDH (h−1) | Total formic acid production (μM) |
|---|---|---|---|---|
| DAV | 24.0 | 1.2 | 18.8 | 120 ± 3.5 |
| AMV | 20.8 | 10 | 16.3 | 130 ± 5.0 |
| CMV | 10.4 | 0.52 | 8.14 | 85.0 ± 1.0 |
| DCV | 8.0 | 0.4 | 6.26 | 83.0 ± 0.9 |
| MV | 12.0 | 0.6 | 9.45 | 101 ± 1.5 |
Next let us focus on the reason for the improvement of visible-light driven CO2 reduction to formic acid with the system of ZnTPPS, 4,4-BPs and FDH. We previously reported that the single-electron reduced form of DAV had high affinity for FDH compared with the other 4,4′-BPs. By enzyme kinetic analysis, the catalytic efficiency of the single-electron reduced form of DAV or AMV was approximately 30 or 2.0 times higher than that of MV and estimated to be more than 560 or 37 times than that of NADH, indicating that 4,4′-BP with an amino group has higher affinity for FDH compared with those of other 4,4′-BPs.43,46,47 From these results, the single-electron reduced form of DAV and AMV was also used as the effective electron carrier for FDH in the visible light-driven CO2 reduction to formic acid. By using DAV and AMV as the electron carrier, the effective visible light-driven CO2 reduction to formic acid with the system consisting of water-soluble zinc porphyrin and FDH in the presence of TEOA was developed.
Conclusions
In this work, the effect of the amino or carboxy-group bonded to 4,4′-BPs on the visible-light driven formic acid production based on CO2 reduction with the system consisting of ZnTPPS and FDH in the presence of TEOA was investigated. In the visible-light driven reduction of 4,4′-BPs with the sensitization of ZnTPPS, the reduction efficiency of 4,4′-BPs depends on the functional ionic group bonded to 4,4′-BPs and effective visible-light driven reduction was observed using 4,4′-BPs with the amino-group, DAV or AMV, compared with those of the other 4,4′-BPs. In the formic acid production from CO2 with the system consisting of TEOA, ZnTPPS, 4,4′-BP and FDH, the formic acid production yield also depends on the functional ionic group bonded to 4,4′-BPs and an effective formic acid production using DAV or AMV was observed compared to the other 4,4′-BPs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by Grant-in-Aid for challenging Exploratory Research (Japan Society for the Promotion of Science) (15K14239), and Grant-in-Aid for Scientific Research on Innovative Areas “Artificial Photosynthesis (2406)”.Notes and references
- J. Kiwi, K. Kalyanasundaram and M. Grätzel, Struct. Bonding, 1982, 49, 37 CrossRef.
- L. A. Kelly and M. A. J. Rodgers, J. Phys. Chem., 1994, 98, 6386 CrossRef CAS.
- J. R. Darwent, P. Douglas, A. Harriman, G. Porter and M. C. Richoux, Coord. Chem. Rev., 1982, 44, 93 CrossRef.
- P. A. Brugger, P. Cuendet and M. Grätzel, J. Am. Chem. Soc., 1981, 103, 2923 CrossRef CAS.
- Y. Amao, Curr. Top. Biotechnol., 2009, 5, 71 CAS.
- J. Kiwi and M. Grätzel, J. Am. Chem. Soc., 1979, 101, 7214 CrossRef CAS.
- Y. Amao, Curr. Nanosci., 2008, 4, 45 CrossRef CAS.
- A. Harriman, G. Porter and M.-C. Richoux, J. Chem. Soc., Faraday Trans. 2, 1981, 77, 833 RSC.
- H. Kumagai, G. Sahara, K. Maeda, M. Higashi, R. Abe and O. Ishitani, Chem. Sci., 2017, 8, 4242 RSC.
- Y. Tamaki and O. Ishitani, ACS Catal., 2017, 7, 3394 CrossRef CAS.
- H. Takeda, C. Cometto, O. Ishitani and M. Robert, ACS Catal., 2017, 7, 70 CrossRef CAS.
- G. Sahara, H. Kumagai, K. Maeda, N. Kaeffer, V. Artero, M. Higashi, R. Abe and O. Ishitani, J. Am. Chem. Soc., 2016, 138, 14152 CrossRef CAS PubMed.
- J. Rohacova and O. Ishitani, Chem. Sci., 2016, 7, 6728 RSC.
- R. Goy, L. Bertini, T. Rudolph, S. Lin, M. Schulz, G. Zampella, B. Dietzek, F. H. Schacher, L. De Gioia, K. Sakai and W. Weigand, Chem. – Eur. J., 2016, 23, 334 CrossRef PubMed.
- G. W. Brudvig, J. N. H. Reek, K. Sakai, L. Spiccia and L. Sun, ChemPlusChem, 2016, 81, 1017 CrossRef CAS.
- Y. Amao, ChemCatChem, 2011, 3, 458 CrossRef CAS.
- I. Okura, Coord. Chem. Rev., 1985, 68, 53 CrossRef CAS.
- I. Okura, Biochimie, 1986, 68, 189 CrossRef CAS PubMed.
- Y. Amao and I. Okura, “Sensitization by Metal Complexes Towards Future Artificial Photosynthesis” in Photocatalysis, Kodansha Springer, 2002 Search PubMed.
- Y. Amao, Y. Maki and Y. Fuchino, J. Phys. Chem. C, 2009, 113, 16811 CAS.
- Y. Zhao, N. C. Anderson, M. W. Ratzloff, D. W. Mulder, K. Zhu, J. A. Turner, N. R. Neale, P. W. King and H. M. Branz, ACS Appl. Mater., 2016, 8, 14481 CrossRef CAS PubMed.
- C. Wombwell, C. A. Caputoand and E. Reisner, Acc. Chem. Res., 2015, 48, 2858 CrossRef CAS PubMed.
- D. Mersch, C.-Y. Lee, J. Z. Zhang, K. Brinkert, J. C. Fontecilla-Camps, A. W. Rutherford and E. Reisner, J. Am. Chem. Soc., 2015, 137, 8541 CrossRef CAS PubMed.
- M. B. Wilker, K. E. Shinopoulos, K. A. Brown, D. W. Mulder, P. W. King and G. Dukovic, J. Am. Chem. Soc., 2014, 136, 4316 CrossRef CAS PubMed.
- E. Reisner, D. J. Powell, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2009, 131, 18457 CrossRef CAS PubMed.
- A. Bachmeier, V. C. C. Wang, T. W. Woolerton, S. Bell, J. C. Fontecilla-Camps, M. Can, S. W. Ragsdale, Y. S. Chaudhary and F. A. Armstrong, J. Am. Chem. Soc., 2013, 135, 15026 CrossRef CAS PubMed.
- M. Ihara, H. Nishihara, K.-S. Yoon, O. Lenz, B. Friedrich, H. Nakamoto, K. Kojima, D. Honma, T. Kamachi and I. Okura, Photochem. Photobiol., 2006, 82, 676 CrossRef CAS PubMed.
- T. Sakai, D. Mersch and E. Reisner, Angew. Chem., Int. Ed., 2013, 52, 12313 CrossRef CAS PubMed.
- C. A. Caputo, L. Wang, R. Beranek and E. Reisner, Chem. Sci., 2015, 6, 5690 RSC.
- Z. Sun, H. Chen, Q. Huang and P. Du, Catal. Sci. Technol., 2015, 5, 4964 CAS.
- D. Dennis and N. O. Kaplan, J. Biol. Chem., 1960, 235, 810 CAS.
- J. Everse and N. O. Kaplan, Adv. Enzymol. Relat. Subj. Biochem., 1973, 37, 61 CAS.
- M. T. Alam, V. Olin-Sandoval, A. Stincone, M. A. Keller, A. Zelezniak, B. F. Luisi and M. Ralser, Nat. Commun., 2017, 8, 16018 CrossRef CAS PubMed.
- Y. V. Rodionov, Uspekhi Microbiologii, 1982, 16, 104 Search PubMed.
- J. G. Ferry, FEMS Microbiol. Rev., 1990, 7, 377 CrossRef CAS PubMed.
- V. I. Tishkov and V. O. Popov, Biomol. Eng., 2006, 23, 89 CrossRef CAS PubMed.
- N. E. Sladek, J. Biochem. Mol. Toxicol., 2003, 17, 7 CrossRef CAS PubMed.
- G. Izaguirre, A. Kikonyogo and R. Pietruszko, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 1997, 118, 59 CrossRef CAS.
- A. Kikonyogo and R. Pietruszko, Biochem. J., 1996, 316, 317 CrossRef CAS PubMed.
- H. Jornvall, B. Persson and J. Jeffery, Eur. J. Biochem., 1987, 167, 195 CrossRef CAS PubMed.
- B. Persson, J. S. Zigler Jr. and H. Jornvall, Eur. J. Biochem., 1994, 226, 15 CrossRef CAS PubMed.
- H. Riveros-Rosas, A. Julian-Sanchez, R. Villalobos-Molina, J. P. Pardo and E. Pina, Eur. J. Biochem., 2003, 270, 3309 CrossRef CAS PubMed.
- R. Miyatani and Y. Amao, Photochem. Photobiol. Sci., 2004, 3, 681 CAS.
- R. Miyatani and Y. Amao, Biotechnol. Lett., 2002, 24, 1931 CrossRef CAS.
- Y. Amao and T. Watanabe, Chem. Lett., 2004, 33, 1544 CrossRef CAS.
- Y. Amao and T. Watanabe, Appl. Catal., B, 2009, 86, 109 CrossRef CAS.
- Y. Amao, Prep. Pap.-Am. Chem. Soc., Div. Fuel Chem., 2010, 55, 91 CAS.
- K. Hironaka, S. Fukuzumi and T. Tanaka, J. Chem. Soc., Perkin Trans. 2, 1984, 1705 RSC.
- R. K. Yadav, G. H. Oh, N.-J. Park, A. Kumar, K.-J. Kong and J.-O. Baeg, J. Am. Chem. Soc., 2014, 136, 16728 CrossRef CAS PubMed.
- R. K. Yadav, J.-O. Baeg, G. H. Oh, N.-J. Park, K.-J. Kong, J. Kim, D. W. Hwang and S. K. Biswas, J. Am. Chem. Soc., 2012, 134, 11455 CrossRef CAS PubMed.
- W. S. Choi, S.-H. Lee, J.-W. Ko and C.-B. Park, ChemSusChem, 2016, 9, 1559 CrossRef CAS PubMed.
- S. K. Kuk, R. K. Singh, D. H. Nam, R. Singh, J.-K. Lee and C.-B. Park, Angew. Chem., 2017, 129, 3885 CrossRef.
- R. Cazelles, J. Drone, F. Fajula, O. Ersen, S. Moldovan and A. Galarneau, New J. Chem., 2013, 37, 3721 RSC.
- X. Ji, Z. Su, P. Wang, G. Ma and S. Zhang, ACS Nano, 2015, 9, 4600 CrossRef CAS PubMed.
- K. T. Oppelt, J. Gasiorowski, D. A. M. Egbe, J. P. Kollender, M. Himmelsbach, A. W. Hassel, N. S. Sariciftci and G. Knör, J. Am. Chem. Soc., 2014, 136, 12721 CrossRef CAS PubMed.
- S. Ikeyama, R. Abe, S. Shiotani and Y. Amao, ACS Catal., 2017 Search PubMed , in submitted.
- D. Mandler and I. Willner, J. Chem. Soc., Perkin Trans. 2, 1988, 997 RSC.
- I. Willner and D. Mandler, J. Am. Chem. Soc., 1989, 111, 1330 CrossRef CAS.
- I. Willner, N. Lapidot, A. Riklin, R. Kasher, E. Zahavy and E. Katz, J. Am. Chem. Soc., 1994, 116, 1428 CrossRef CAS.
- I. Willner, I. Willner and N. Lapidot, J. Am. Chem. Soc., 1990, 112, 6438 CrossRef CAS.
- R. Miyatani and Y. Amao, Biotechnol. Lett., 2002, 24, 1931 CrossRef CAS.
- R. Miyatani and Y. Amao, J. Mol. Catal. B: Enzym., 2004, 27, 121 CrossRef CAS.
- R. Miyatani and Y. Amao, J. Jpn. Pet. Inst., 2004, 47, 27 CrossRef CAS.
- Y. Amao, R. Abe and S. Shiotani, J. Photochem. Photobiol., A, 2015, 313, 149 CrossRef CAS.
- I. Tsujisho, M. Toyoda and Y. Amao, Catal. Commun., 2006, 7, 173 CrossRef CAS.
- M. Kodaka and Y. Kubota, J. Chem. Soc., Perkin Trans. 2, 1999, 891 RSC.
- Y. Amao, Chem. Lett., 2017, 46, 780 CrossRef.
- Y. Amao and S. Ikeyama, Chem. Lett., 2015, 44, 1182 CrossRef CAS.
- S. Ikeyama and Y. Amao, Chem. Lett., 2016, 45, 1259 CrossRef CAS.
- S. Ikeyama and Y. Amao, ChemCatChem, 2017, 9, 833 CrossRef CAS.
- S. Ikeyama, R. Abe, S. Shiotani and Y. Amao, Chem. Lett., 2016, 45, 979 CrossRef.
- S. Ikeyama and Y. Amao, Sustainable Energy Fuels, 2017, 1, 1730 CAS.
- N. Kato, H. Sahm and F. Wagner, Biochim. Biophys. Acta, 1979, 566, 12 CrossRef CAS.
- N. L. Allinger, J. Am. Chem. Soc., 1977, 99, 8127 CrossRef CAS.
- M. Kaneko, K. Suzuki, E. Ebel and D. Wöhrle, Macromol. Symp., 2003, 204, 71 CrossRef CAS.
- Y. Amao and I. Okura, J. Mol. Catal. A: Chem., 1996, 105, 125 CrossRef CAS.
- S. Karastogianni and S. Girousi, Sensing in Electroanalysis, ed. K. Kalcher, R. Metelka, I. Švancara and K. Vytřas, University Press Centre, Pardubice, Czech Republic, 2013/2014, vol. 8, pp. 241–252 Search PubMed.
- K. Kalyanasundaram and M. Nerman-Spallart, J. Phys. Chem., 1982, 86, 5163 CrossRef CAS.
- J. M. Lehn and J. P. Sauvarge, Nouv. J. Chim., 1977, 1, 449 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7pp00277g |
| This journal is © The Royal Society of Chemistry and Owner Societies 2018 |