
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rationalisation of a mechanism for sensing single point variants in target DNA using anthracene-tagged base discriminating probes†
Jean-Louis H. A.
Duprey
a,
Dario M.
Bassani
 b,
Eva I.
Hyde
c,
Gediminas
Jonusauskas
d,
Christian
Ludwig
e,
Alison
Rodger
f,
Neil
Spencer
a,
Joseph S.
Vyle
g,
John
Wilkie
a,
Zheng-Yun
Zhao
a and
James H. R.
Tucker
*a
b,
Eva I.
Hyde
c,
Gediminas
Jonusauskas
d,
Christian
Ludwig
e,
Alison
Rodger
f,
Neil
Spencer
a,
Joseph S.
Vyle
g,
John
Wilkie
a,
Zheng-Yun
Zhao
a and
James H. R.
Tucker
*a
aSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK. E-mail: j.tucker@bham.ac.uk
bInstitut des Sciences Moléculaires, CNRS UMR 5255, Université Bordeaux, 351 Cours de la Libération, Talence 33405, France
cSchool of Biosciences, The University of Birmingham, Edgbaston, Birmingham B15 2TT, UK
dLaboratoire Ondes et Matière d'Aquitaine, UMR CNRS 5798, Université Bordeaux, Talence 33405, France
eHenry Wellcome Building for Biomolecular NMR Spectroscopy, Institute of Cancer & Genomic Sciences, College of Medical & Dental Sciences, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK
fDepartment of Molecular Sciences, Faculty of Science and Engineering, Macquarie University, North Ryde, NSW 2109, Australia
gSchool of Chemistry and Chemical Engineering, Queen's University Belfast, David Keir Building, Stranmillis Road, Belfast, BT9 5AG, UK
First published on 15th August 2018
Abstract
The labelling of DNA oligonucleotides with signalling groups that give a unique response to duplex formation depending on the target sequence is a highly effective strategy in the design of DNA-based hybridisation sensors. A key challenge in the design of these so-called base discriminating probes (BDPs) is to understand how the local environment of the signalling group affects the sensing response. The work herein describes a comprehensive study involving a variety of photophysical techniques, NMR studies and molecular dynamics simulations, on anthracene-tagged oligonucleotide probes that can sense single base changes (point variants) in target DNA strands. A detailed analysis of the fluorescence sensing mechanism is provided, with a particular focus on rationalising the high dependence of this process on not only the linker stereochemistry but also the site of nucleobase variation within the target strand. The work highlights the various factors and techniques used to respectively underpin and rationalise the BDP approach to point variant sensing, which relies on different responses to duplex formation rather than different duplex binding strengths.
Introduction
There is ongoing interest in developing cheap and effective methods for reading out single nucleobase changes (point variants) in the human genome due to their association with diseases with a genetic component. These variants include single nucleotide polymorphisms (SNPs),1 defined as variations at specific base positions within the genome in >1% of the population, as well as cancer-causing mutations2 that occur over the life-time of an individual.3 Current fluorescence sensing methods (in particular commercial ones) for identifying point variations within a sequence of sample DNA typically use a hybridization (i.e. duplex forming) approach‡ that relies on differences in thermodynamic stability between a probe–target duplex that is fully complementary and one that bears a base pair mismatch at the variant site.4,5 The desired difference in output signal between two given target strands thus relies on just one of them forming a stable duplex with a fluorescent probe under the assay conditions. This leads to the task of identifying, for each new sequence, a temperature window in which the assay can work effectively.An alternative approach to point variant sensing is to generate different and distinct read-outs for the two possible duplexes formed with the probe strand, as illustrated for this work in Fig. 1. From a sensor design point of view, such an approach is attractive since it removes the need to establish a temperature window in which one target strand binds and the other does not. This means that sensing can be carried out at any desired temperature, so long as each target can form a stable duplex. Although commercial assays of these so-called base discriminating probes§ (BDPs) have yet to emerge, there are plenty of examples from the research literature over the last few years,6–19 with read-out methods other than fluorescence also recently established.20
 | ||
| Fig. 1 Schematic representation of the point variant DNA sensing mechanism relevant to this study. An anthracene-tagged probe strand with an L-threoninol linker discriminates between two DNA target strands that differ in sequence by a single base change through either (a) a decrease – matching base-pair (CG), or (b) an increase – mismatching base-pair (shown as CA), in fluorescence emission intensity upon duplex formation. | ||
Our main contribution to this area has been the development of anthracene-containing probes in which the tag is attached to the DNA backbone via the non-nucleosidic linkers serinol17 or threoninol.11,15,16 These linkers are particularly attractive due their relative ease of preparation and, in the case of threoninol, the ready availability of both the D- and L-isomers.21 While others have developed similar strands containing pyrene12,18 and thiazole orange10,13,19 tags, anthracene is attractive due to its well documented intercalative30 and photochromic22,38 properties. Our previous work established that the formation of duplexes with fully matched base pairs on either side of the anthracene tag brought about a decrease in emission intensity, whereas the formation of those containing a base-pair mismatch on the 5′ side with respect to the tag brought about an increase (Fig. 1).16,17 Although these observations have proven to be broadly applicable to different sequences,16 making this sensing method attractive for probing almost any nucleobase variation, there has been no in-depth rationalisation of this intriguing OFF/ON behaviour, without which it cannot be deemed a robust transferable approach. Herein, through a combination of spectroscopic techniques and molecular dynamics simulations, we provide an explanation for the sensing mechanism. In particular, we show how both the choice of the linker group stereochemistry as well as the position of the mismatched base-pair with respect to the fluorophore are essential for generating an effective sensing response. This study is important for highlighting the various factors that underpin the design of a successful base discriminating probe, which in turn should inform the design of the most effective probes of this type for commercial or clinical applications.
Results
Synthesis and characterisation
The anthracene tag monomers for DNA incorporation were prepared in three steps from the known anthracene carboxylic acid 1,23 which was first reacted with D- or L-threoninol respectively, followed by DMT protection and then phosphitylation to form the corresponding phosphoramidite (Scheme 1). Standard automated DNA synthesis incorporated these tags into the middle of two 15-mer sequences (Table 1).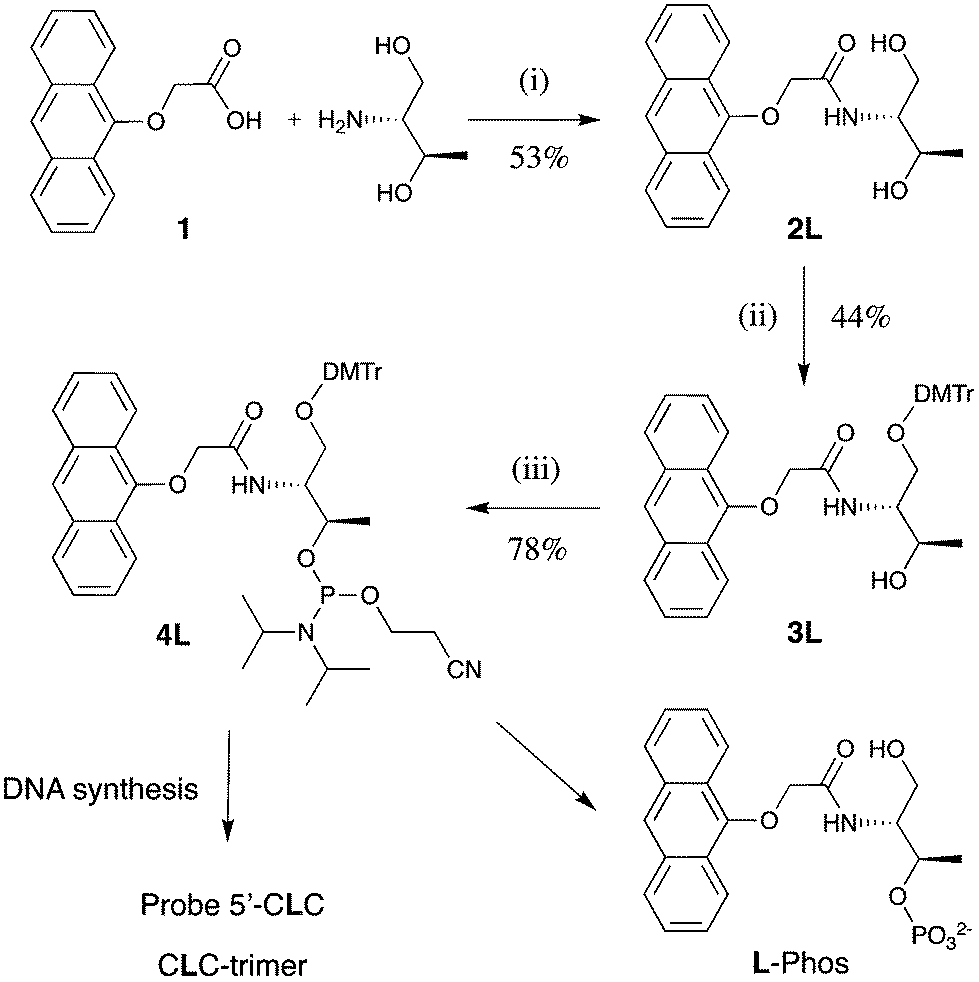 | ||
| Scheme 1 Synthetic route followed to the anthracene-tagged strands, shown for the L-threoninol linker. Conditions: (i) HOBt, DIPC, DMF, 12 h, 40 °C; (ii) DMTCl, pyridine, 12 h, RT; (iii) C9H18N2OClP, DCM, 30 min, RT; L-Phos is the control monomer. | ||
| Oligonucleotide name | Sequence |
|---|---|
| a B = A, C, G, T or Ab (Ab denotes an abasic nucleoside with no nucleobase, prepared for the 3′-GBG strand only). | |
| 5′-CTC | 5′-TGGACT-CTC-TCAATG-3′ |
| Probe 5′-CXC | 5′-TGGACT-CXC-TCAATG-3′ |
| Target 3′-GBG | 3′-ACCTGA-GBG-AGTTAC-5′ |
| Target 3′-GAB | 3′-ACCTGA-GAB-AGTTAC-5′ |
| Target 3′-BAG | 3′-ACCTGA-BAG-AGTTAC-5′ |
| CLC-trimer | 5′-CLC-3′ |
Notwithstanding that the sensing behaviour operates for other flanking bases,16 for the sake of comparison with most of our previous work, it was decided to retain two cytosines either side of the anthracene tag for this detailed study, giving the 15-mer probes 5′-CLC- and 5′-CDC (Table 1). In addition, three control compounds were made: the unmodified strand 5′-CTC, the 3-mer CLC-trimer and the monomer L-Phos. So that we could investigate and rationalise the effect of variations in the target DNA sequence on both the thermal stabilities and the photophysical properties of various duplexes, a series of 15-mer target sequences (3′-GBG, 3′-GAB, 3′-BAG where B = G, A, C, T) were also prepared. In particular, this would allow us to examine the effect of introducing changes in the adjacent 5′- (upstream) or 3′- (downstream) positions as well as those directly opposite the tag. Purification was performed via preparative RP-HPLC on the 5′-DMT protected strands, which were then detritylated and de-salted. The purity and composition of each oligonucleotide was confirmed using analytical HPLC and mass spectrometry respectively, as detailed in the ESI.†
Fluorescence spectroscopy
Upon excitation of the anthracene moiety at 350 nm, 15-mer probes 5′-CDC and 5′-CLC were both found to give a structured emission band, characteristic of anthracene, with a maximum at 422 nm (Fig. 2). Interestingly, the CDC system was much more emissive, with a quantum yield (Φ = 0.21) more than double that of the CLC probe (Φ = 0.09) and approaching that of the anthracene phosphate monomer L-Phos (Φ = 0.28). The fluorescence response profiles of all the duplexes were then evaluated by determining changes in the anthracene fluorescence emission intensity upon duplex formation (measured as a percentage change in emission intensity compared to the single strand at 426 nm). These data, along with various fluorescence quantum yields, are presented in the ESI,† with representative spectra presented in Fig. 2. Upon the addition of the fully matching 3′-GAG strand to probes 5′-CLC and 5′-CDC (matching in the sense that all probe nucleobases are complementary to those of the target), a decrease in emission intensity was observed in each case (−75% and −72% respectively at 426 nm), with the spectra becoming somewhat broader and less well defined. This effect was observed for all the fully matching systems studied, irrespective of the identity of the nucleobase directly opposite the tag or the stereochemistry of the linker. Of particular relevance for base sensing purposes were the results for the mismatched duplexes: once again, decreases in fluorescence intensity were observed for all the 5′-CDC duplex systems. However for the 5′-CLC probe, whereas mismatches immediately downstream from the tag site (e.g. 3′-GAA) also led to a decrease, upstream mismatches led to a significant increase in intensity. In the case of 3′-AAG, the increase was +86% at 426 nm (Fig. 2). These results indicate that the stereochemistry of the linker within these CXC probes is a determining factor in generating effective point variant sensing, resulting in what is essentially an OFF/ON signal response for the CLC system. | ||
| Fig. 2 Fluorescence spectra showing the change in emission upon hybridisation of anthracene Probes 5′-CXC, with Target 3′-GAG and Target 3′-AAG (top row) and Targets 3′-AAG and 3′ GAA (bottom row) λex 350 nm, 1 μM probe concentration, 10 mM pH 7 phosphate buffer, 100 mM NaCl, room temperature. | ||
Fluorescence lifetimes
Fluorescence lifetimes for the anthracene phosphate monomer L-Phos, the anthracene CLC-trimer, the two 15-mer probe strands and their duplexes revealed some interesting trends. The decay for L-Phos was found to be mono-exponential (τ = 5.1 ns) and the trimer CLC bi-exponential (τ1 = 1 ns and τ2 = 2.5 ns). For the two 15-mer probes and all their duplexes, a third longer-lived pathway was also identified, with each sample accordingly having a short (τ1), a medium (τ2) and a long (τ3) component, suggesting a minimum of three anthracene environments that are not rapidly interconverting.24 Multiple decay rates have previously have been noted for pyrene,25 acridine26 and thiazole orange27 fluorophores when tagged to oligonucleotides. All the data are presented in the ESI,† with a selection presented in Table 2.| Sequence | τ 1 wt (ns) (%) | τ 2 wt (ns) (%) | τ 3 wt (ns) (%) | χ 2 | |||
|---|---|---|---|---|---|---|---|
| a Relative contribution of each decay constant to the total emission decay. | |||||||
| 5′-CLC | 1.05 | (38) | 4.28 | (36) | 12.4 | (26) | 1.04 |
| 3′-GAG | |||||||
| 5′-CDC | 1.50 | (33) | 3.68 | (41) | 9.08 | (25) | 0.99 |
| 3′-GAG | |||||||
| 5′-CLC | 0.94 | (32) | 4.08 | (40) | 12.9 | (28) | 1.01 |
| 3′-GAA | |||||||
| 5′-CLC | 0.88 | (2) | 4.6 | (21) | 14.4 | (77) | 0.97 |
| 3′-AAG | |||||||
| 5′-CLC | — | 5.1 | (20) | 13.3 | (80) | 1.02 | |
| 3′-CAG | |||||||
| 5′-CLC | — | 5.9 | (29) | 13.5 | (71) | 0.97 | |
| 3′-TAG | |||||||
The effect on the lifetime distribution of the mismatched duplexes was also investigated. Those formed with the 5′-CDC probe were again all tri-exponential with comparable decay profiles. However, whereas duplexes for the 5′-CLC system with downstream mismatches (e.g. 3′-GAA) also gave tri-exponential decays, those for upstream mismatches changed from a tri-exponential decay profile to essentially a bi-exponential one, with the fastest quenching pathway absent (or almost completely suppressed, only 2%, in the case of 3′-AAG).
Transient absorption spectroscopy
To investigate whether ultrafast (i.e. sub-nanosecond) processes were involved in the quenching of the anthracene excited state and, if so, whether these included any oxidative or reductive processes, femtosecond transient absorption spectroscopy (TAS) experiments were performed on the matched 5′-CLC/3′-GAG and mismatched 5′-CLC/3′-AAG duplexes. These samples were available in sufficient quantities from the NMR studies (vide infra). The femtosecond TAS data is available in the ESI.† In both cases, an absorption band (λmax = 580 nm) was observed immediately following the excitation pulse (λex = 365 nm, 50 fs pulse). This band is attributed to the Sn ← S1 absorption of the excited singlet state based on its similarity in shape and energy to that observed for 9-substituted anthracenes.28 For the matched duplex, the decay of the excited singlet occurred with a sub-nanosecond decay component (τ = 260 ps) and was accompanied by a very small rise in the signal at ca. 690 nm (no distinct new absorption band could be detected). The rise component of this long-wavelength transient at 690 nm was very fast (0.4 ps) and could be indicative of the inefficient formation of anthracene radical ions whose absorbance maximum is expected at 650–750 nm.29 In contrast, no transient absorption signal was detected at these longer wavelengths in the case of the mismatched duplex, whose S1 state decayed with a rate that exceeded the time resolution of our instrument (3.5 ns).UV/vis spectroscopy and melting temperature measurements
Absorbance spectra of the single stranded anthracene probe 5′-CLC gave a 5 nm red shift in the λmax for the S0 → S1 (330–400 nm) band compared to that for the corresponding anthracene monophosphate L-Phos, with a further red shift and a hypochromic effect observed upon duplex formation with 3′-GAG (ESI†). These effects indicate the existence of electronic interactions between the anthracene and adjacent proximate nucleobases.10,30 The thermal stability of each duplex was determined in the usual way using UV/Vis spectroscopy by monitoring absorbance changes at 260 nm as a function of temperature (see Table 3 and ESI†). The matching 5′-CLC/3′-GAG duplex gave a higher Tm value (52.5 °C) than the corresponding duplex with the 5′-CDC probe (48 °C), indicating a greater stabilising effect on the duplex when the anthracene is connected via the L-threoninol linker. Changing the base opposite the anthracene on the target strand (Targets 3′-GBG) had no significant effect on this difference (Tm range 52.5–51 °C for 5′-CLC duplexes and 46–48 °C for 5′-CDC duplexes). Notably, the control duplex 5′-CTC/3′-GAG gave a Tm value of 55 °C, indicating a much smaller decrease in melting temperature upon replacing a H-bonding T base with the L-threoninol linked anthracene. Interestingly, the removal of the A base opposite the anthracene (3′-GAbG) resulted in a slightly higher Tm value for the 5′-CLC system, suggesting that the anthracene can provide further stabilisation when a cavity opposite the tag is present. As expected, decreases in Tm values were observed on going from matched to mismatched duplexes31 but, importantly for the 5′-CLC system, the decrease upon introducing mismatches (max 8 °C) was less marked than it was for the 5′-CDC system (max 13 °C) or for the unmodified 5′-CTC system (max 15 °C). This indicates an additional stabilising influence from the anthracene unit in the mismatched duplex, but only when connected to the L-threoninol linker.| Target | ||||
|---|---|---|---|---|
| a Average of at least 3 runs and determined by taking the maximum of the first derivative of the melting curve. | ||||
| Probe | 3′-GAG | 3′-GAA | 3′-AAG | 3′-GAbG |
| 5′-CTC | 55.0 | 42.5 | 42.0 | 40.0 |
| 5′-CLC | 52.5 | 44.5 | 46.0 | 53.5 |
| 5′-CDC | 48.0 | 37.5 | 35.0 | 48.0 |
Circular dichroism spectroscopy
In order to examine the structural form of the modified DNA and to study the interactions between anthracene and the neighbouring bases, CD spectra of various unmodified and modified duplexes were recorded. All three fully matching duplexes adopted the expected B-DNA structure (Fig. 3), but with a notable shoulder at ca. 250 nm observed for the modified duplexes, which was more apparent for the 5′-CLC system. This can be explained by an induced CD signal arising from the anthracene chromophore (S0 → S2 band) upon its interaction with the DNA base stack.32,33 A more concentrated sample also revealed an induced CD signal for the lower energy S0 → S1 band (Fig. 3 inset). Differences between the CD spectra for the anthracene signals in the matched and various mismatched duplexes (see ESI†) highlight the effectiveness of this technique in indicating subtly different positions for the anthracene tag in its interaction with the duplex backbone. | ||
| Fig. 3 CD spectra of duplexes 5′-CTC/3′-GAG, 5′-CLC/3′-GAG and 5′-CDC/3′-GAG in 10 mM pH 7 phosphate buffer, 100 mM NaCl. Main: [duplex] = 5 μM; insert: S0 → S1 region, [duplex] = 500 μM. | ||
NMR studies
In order to gain further information on how the anthracene tag was affecting the structures of the duplexes, 500 MHz 1H NMR spectra were run on the unmodified DNA duplex, both matching duplexes (5′-CLC/3′-GAG and 5′-CDC/3′-GAG) and the mismatched 5′-CLC/3′-AAG system. Measurements were carried out in buffered D2O at a relatively high duplex concentration of ca. 1 mM. Initially, the anthracene peaks were assigned from an NMR spectrum of the uncoupled anthracene in phosphate monomer L-Phos. These were then compared to a spectrum of the CLC-trimer in order to evaluate the effect of surrounding bases (see ESI†), which revealed strong upfield shifts of between 0.25 and 0.52 ppm for the five anthracene proton signals. TOCSY and COSY experiments were then used to identify the same protons on the 5′-CLC/3′-GAG and 5′-CLC/3′-AAG systems, which revealed further upfield shifts for these protons, which is explained by their proximity to, and interaction with, the base stack within dsDNA.The imino regions in each NMR spectrum, denoting the H-bonding base pairs (N–H⋯N resonances, where the proton donor group is from either a T or G base), are shown in Fig. 4. The unmodified duplex (Fig. 4a) shows a cluster of peaks in the A-T region and six peaks further upfield in the G-C region, indicating the loss of one terminal GC base pair signal out of the possible seven due to fraying of the duplex ends. The spectra for the anthracene-modified duplexes show interesting trends. For the two matching duplexes, distinct upfield shifts are observed for two of the G-C signals, which are assigned to NH protons on each G base lying either side of the modification site. In the case of the L-threoninol system, these signals are distinct from one another at 11.20 ppm and 12.15 ppm. For the mismatched duplex, and as expected from these assignments, one upfield peak is no longer observed (due to the introduction of a G-A mismatch) with one residual peak now at 11.9 ppm.
 | ||
| Fig. 4 500 MHz 1H NMR spectra of (a) 5′-CTC/3′-GAG (b) 5′-CDC/3′-GAG, (c) 5′-CLC/3′-GAG and (d) 5′-CLC/3′-AAG. Recorded at 27 °C, 5% D2O in H2O, [duplex] = 950 μM, 10 mM pH 7 phosphate buffer, 100 mM NaCl. | ||
Molecular dynamics modelling
The considerable differences in the spectroscopic and thermodynamic properties of the duplexes as a function of tag stereochemistry and target sequence encouraged us probe their structures further using molecular dynamics (MD) simulations. Calculations on the matching 5′-CLC/3′-GAG, 5′-CDC/3′-GAG and mismatching 5′-CLC/3′-AAG duplexes were undertaken using the AMBER 11 package (see ESI† for more details). The results indicate that the two threoninol stereoisomers position the anthracene in different environments (Fig. 5), with L-threoninol tag able to intercalate with the duplex via the major groove, whereas a minor groove position, away from the base-pair stack, is favoured for the D-threoninol tag. The results for the mismatched duplex 5′-CLC/3′-AAG show a greater variation in the position of the tag during the dynamics runs, as well as a trend for the anthracene to insert itself more deeply inside the DNA duplex (Fig. 6). | ||
| Fig. 5 Images from AMBER molecular dynamics models of 5′-CLC/3′-GAG duplex (left) and 5′-CDC/3′-GAG duplex (right) after 10 ns. The carbon atoms of the anthracene monomer unit are coloured yellow. | ||
 | ||
| Fig. 6 Images from AMBER molecular dynamics models of matching 5′ CLC/3′-GAG duplex (left) and mismatching 5′ CLC/3′-AAG duplex (right) after 10 ns. The carbon atoms of the anthracene monomer unit are coloured yellow, the nucleobases to the 3′ side of the anthracene are coloured blue and the bases to the 5′ side are coloured red (mismatched adenine in target strand denoted with an arrow). | ||
Discussion
The chiral threoninol group is a popular choice as a means of attaching functional tags to the backbone of DNA, with examples of connections made to nucleobases,34 azobenzene,35 psoralen,36 acridine26,37 and anthracene.15–17,21,38 Despite this, relatively few studies have investigated in depth the effects of changing the linker stereochemistry on the properties and role of the DNA-appended tag. Our results indicate a remarkable stereochemical dependence on both the photophysical and thermodynamic properties of the duplexes. This dependence results in the DNA probe with the L-linker being the much more effective point variant sensor for this tether length, with the signal decreasing when a fully matched duplex is formed but increasing for one containing just one base pair mismatch (Fig. 1). However, as well as stereochemistry being a major factor, this effect is highly dependent on the site of base variation within the target sequence, with only a change upstream of the anthracene (e.g. 3′-![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) AG to 3′-
AG to 3′-![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) AG) giving a significant discriminatory (OFF/ON) effect. Taken as a whole, our studies indicate that the position and orientation of the anthracene tag plays a crucial role in explaining this phenomenon, as discussed below.
AG) giving a significant discriminatory (OFF/ON) effect. Taken as a whole, our studies indicate that the position and orientation of the anthracene tag plays a crucial role in explaining this phenomenon, as discussed below.
If we first consider the single stranded probes, the quantum yield for the D-threoninol linker system is more than twice as high as for its L-threoninol counterpart, with a value fairly close to that of the anthracene monomer alone (L-Phos). This indicates that with a D stereochemistry, the tag is positioned such that its excited state is much less sensitive to quenching processes attributed to vicinal bases. However, upon duplex formation, the anthracene necessarily comes into contact with more bases, which for both stereochemistries and for almost all sequence variations, with the one noted exception, decreases the emissiveness of the anthracene tag. This effect is not surprising, given that DNA bases are known to quench the excited states of various organic fluorophores.25 However, it is worth noting that the anthracene quenching is uniform when varying the base opposite the tag for both the L-system and D-systems (i.e. Probe 5′-CLC with targets 3′-GBG, where B = G, A, C and T, see ESI†) and this trend continues for an abasic site at this position. This gives a strong indication that the base directly opposite the anthracene tag plays no direct role in quenching its excited state. This contrasts with probes with longer tether lengths between the anthracene and the L- or D-threoninol backbone, for which either base variations15 or base modifications (i.e. epigenetic changes)11,15 may be sensed by changes in fluorescence emission intensity.
As noted above, the one notable exception to the general trend of anthracene emission intensity decreasing upon duplex formation is for the L-system, but only when duplexes are formed that contain a base-pair mismatch immediately upstream of the tag. Furthermore the effect is observed for both transversions and transitions (i.e. an increase in emission is observed for all mismatched duplexes 5′-CLC/3′-BAG, where B = A, C, T). Our spectroscopic measurements and molecular simulations indicate that the origin of this effect lies in the extent to which a given stereochemistry enables the anthracene tag to experience a local environment that is less quenching upon the introduction of a mismatching base pair. For example, the data from the variable temperature UV/vis studies (Table 3) indicates that of the two isomers, the L stereochemistry confers substantially more stabilisation than the D in the matching systems, suggesting a greater interaction between the tag and the duplex. For the mismatched duplexes, there is an expected drop in the duplex melting temperature for the unmodified and the modified systems with either stereochemistry. However, whereas the location of the mismatch (upstream or downstream) does not appear to substantially affect this change in stability, the smaller drop in Tm values for the L-isomer systems indicates that this stereochemistry allows the anthracene to insert further into the mismatched duplexes, providing greater hydrophobic stacking interactions that hold them together more strongly in the absence of local hydrogen bonding.
CD spectroscopy also reveals an interesting stereochemical effect (Fig. 3). The characteristic B-DNA structure is observed for the matching duplexes, with additional anthracene-dependent signals that are attributed to be a result of induced CD (ICD).39 These signals arise due to the transfer of chirality to the chromophore and are expressed as a positive Cotton effect at ca. 260 nm (S0 → S2). The ICD signals are most likely due to excitonic coupling between the anthracene and proximate bases and indicate different electronic environments experienced by the tag in various duplexes. The less intense peak for the D-isomer system would again be consistent with this stereochemistry leading to a weaker interaction with the base pair stack. However, the orientation of the anthracene is also an important consideration. The position in which the long axis is at 45° to that of the adjacent base pairs is expected to give the strongest ICD signal, compared to an angle closer to 0 or 90°. The spectrum of concentrated samples reveals an additional CD band in the 330–420 nm region (S0 → S1) which is stronger for the L-isomer duplex, again indicating a strong interaction with the base pair stack. This band has previously been observed in untethered anthracene groups that intercalate into DNA24,40 and indicates a binding mode where the anthracenyl short axis can align relative to the base pair stack.41,42 The S0 → S2 anthracene band (at ∼ 260 nm) in spectra of the mismatched 5′-CLC duplexes (see ESI†) is clearly still prominent, which again can be explained by significant interactions with the bases. However its broadening suggests that the tag can now adopt a greater number of conformations in the more loosely held duplex.
The 1H NMR spectra of the duplexes (Fig. 4) also support the existence of different local environments for the anthracene tag that affect their sensing properties. For both matching duplexes (5′-CLC/3′-GAG and 5′-CDC/3′-GAG), the two signals for the imino protons associated with the GC base pairs directly adjacent to the anthracene tag are shifted upfield compared to the unmodified control duplex, with the largest change observed in the L-linker system. While this can be explained by weaker H-bonding interactions and a greater exchange with the solvent, intercalation and stacking can also induce upfield shifts due to the ring current effect of the chromophore.43 Based on the molecular modelling studies showing a better orbital overlap of the anthracene with the upstream base (with respect to the tag) and a shorter distance of approach (vide infra), this more upfield-shifted signal is ascribed to the 3′-AG imino proton. For the mismatching 5′-CLC/3′-AAG system, it is interesting to note that the remaining H-bonded imino G signal (i.e. 3′-AA) is shifted further upfield than the equivalent base in the 3′GAG duplex, which is consistent with a greater degree of duplex insertion for the tag in the mismatched system, in support of the proposed model.
The lifetime data in Table 2 provide access to quenching rates of the excited anthracene tag, which are important for elucidating the sensing mechanism. The presence of multiple lifetimes in oligonucleotide conjugated fluorophores is not unusual and has been studied previously using 2-aminopurine (2AP) nucleobase,44 or nucleobase-appended BODIPY45 and PNA conjugated thiazole orange.27 The multiple decay parameters present in our data indicate a dynamic profile for the anthracene in which it partitions between different environments in its interaction with the duplex. These different environments are not interconverting within the lifetime of the excited states. As would be expected, the monomer L-Phos emits with a single lifetime (5.1 ns) that is somewhat shorter than that of the chromophore in a non-aqueous environment, due to quenching by water. Based on this, we attribute the 4 ns decay component (τ2) observed in all the ss and dsDNA species to the anthracene chromophore located in a predominantly aqueous environment. In the case of the L-isomer, this would correspond to the anthracene adopting an extra-helical conformation, while for the D-isomer, a minor groove conformation is also possible, according to the molecular modeling simulations (Fig. 5).
The fluorescence lifetime measurements and the time-resolved absorption data respectively indicate the presence of additional fast (ca. 1 ns) and ultrafast (ps) quenching processes in the matching 5′-CLC/3′-GAG system. Furthermore it is clear that the point variant sensing mechanism, triggered by the conversion of a matching to a mismatching base pair immediately upstream of the tag, is accompanied by the disappearance of both of these processes. In assigning these processes, we note the absence of any transient species on the nanosecond timescale ascribable to oxidized or reduced anthracene, or to the population of the anthracene triplet state. This effectively rules out the 1 ns decay process (assigned to τ1 in Table 2) being due to photoinduced electron transfer (PET) that forms the corresponding anthracene radical ion (or population of the triplet state through charge recombination following PET). Instead, it most likely corresponds to fast (8.5 × 108 s−1) internal conversion (IC) of the S1 state as a result of collisional quenching by the close proximity of neighbouring bases. It is worth noting that the lifetime measurements reveal that at least one of these fast decay pathways assigned to the τ1 decay component is also present in both ssDNA probes as well as the CLC-trimer (see ESI†). This suggests that quenching is not solely dependent on the presence of bases in the target strand, but rather on the tightness of the immediate cavity of adjacent bases in the same strand that frame the tag.
An ultrafast (ps) decay process, such as that observed for the 5′-CLC/3′-GAG duplex, could provide an explanation for the similarity in fluorescence lifetimes between the D and L systems (both unbound and bound as fully matched complexes) despite having very different quantum yields. However the question arises as to whether this picosecond decay is also due to collisional interactions or alternatively a PET process. There is indeed evidence for the formation of an anthracene radical ion whose transient decay is detected at 690 nm (see ESI†). However the increase in signal intensity is weak and its rate of formation (k ≈ 2.5 × 1012 S−1) is in fact faster than that of the ultrafast decay component of the S1 state (270 ps). Therefore it would appear that collisional quenching by proximate bases is the most likely explanation for both the ultrafast and fast decay processes of the S1 state, which at least in the case of the 5′-CLC/3′-GAG system, start in the picosecond domain and continue into the low nanosecond domain. However the fact that this additional ultrafast electron transfer process is also absent in the mismatched 5′-CLC/3′-AAG system might suggest that the guanine base in the upstream position from the anthracene tag in the matched target (i.e. 3′-AG) is involved in this process through its oxidation.26 This would thereby result in an increase in fluorescence emission intensity by its removal (i.e. the formation of a mismatch). However, such a reliance on the presence or absence of guanine in the target strand for the observed base variant sensing behaviour would be at odds with our previous studies16 that have demonstrated the same mismatch-induced sensing effects when targeting C-to-A and T-to-A variations (i.e. targets not involving pairing with a guanine base in the matching target strand).
The molecular dynamics models are useful for visualising the environment around the anthracene in terms of how it might effect possible decay pathways for the excited state. For example, the cavity around the anthracene widens to some extent in the mismatched system (Fig. 6), which could account for the absence of the τ1 pathway involving collisional quenching by proximate base pairs. Another important consideration in this respect is the third and longest decay component, τ3 (range 9–14 ns). The wider cavity would give more room for the tag to insert itself further into the duplex, placing it into a more hydrophobic environment, where it is protected from quenching water molecules. In fact, this decay parameter is certainly similar to the lifetime of 9-alkoxyanthracene derivatives in non-polar organic solvents (7 ns).46 The absence of τ3 in the CLC trimer (ESI†) and guanine tagged anthracenes47 is in agreement with this hypothesis, as these conjugates have more open structures that would preclude conformations in which quenching processes from water and proximate bases could be avoided.
The question as to why only a change to the upstream position (with respect to the tag, i.e. 3′-AG) gives rise to the sensing effect can most likely be explained in terms of the anthracene-base distance and/or overlap. Once again, the molecular dynamics simulations are informative as they consistently show a better orbital overlap of anthracene with this base and a shorter distance of approach.32 They also shed considerable light on the importance of the threoninol linker stereochemistry. In the case of the matching duplex 5′-CLC/3′-GAG, the stereochemistry of the linker allows the anthracene to intercalate into the centre of the duplex via the major groove (Fig. 5), to the extent that the tag can locate itself in approximately the same position as a natural nucleobase. As such, it benefits from considerable overlap with the adjacent nucleobases and the resulting stabilisation through hydrophobic pi-stacking interactions.48 At the same time it does not clash with the adenine opposite and allows a natural, un-kinked structure to be maintained by the duplex. Hence, the melting point for this duplex is almost as high as that of unmodified DNA (Table 3). In contrast for the matching duplex with the D-threoninol isomer, the stereochemistry is such that duplex intercalation is much less favoured (Fig. 5, see also ESI†), with the anthracene instead orientating itself towards the minor grove, leading to a lower melting point. For this duplex, the models indicate that the adenine directly opposite the tag follows suit by flipping out of the duplex, presumably to maximise the stacking interactions between the residual H-bonding base pairs either side of the modification site.
Conclusions
Through a combination of detailed photophysical studies, NMR studies and molecular modelling, we have been able to rationalise the sensing mechanism of anthracene-tagged DNA probes that detect single nucleobase changes (point variants) in target DNA strands. The sensing process, involving changes in anthracene fluorescence emission intensity upon duplex formation, is highly dependent upon the stereochemistry of the tag linker as well as the site of the mismatch. Taken together, the data suggests that the preferred OFF/ON signal observed with the L-threoninol linker can be chiefly attributed to this stereochemistry enabling a close intercalative interaction between the tag and the duplex stack. This allows the anthracene to experience a different environment within a mismatched duplex, with room for it to be further embedded inside the duplex and consequently more shielded from quenching pathways triggered by flanking bases and water. This work highlights the various factors that need be considered in the design of an effective base discriminating probe (BDP) as well as the techniques that can be used to explain its mode of operation. It also illustrates how substantially different photophysical properties of a short strand of functionalised DNA can arise as a consequence of just one small stereochemical modification.Conflicts of interest
There are no conflicts to declare.Acknowledgements
JHRT acknowledges The Centre for Chemical and Materials Analysis in the School of Chemistry for technical support and the EPSRC for the award of a Leadership Fellowship (EP/G007578/1).References
- (a) K. Nakatani, ChemBioChem, 2004, 5, 1623–1633 CrossRef PubMed; (b) A. L. Price, C. C. A. Spencer and P. Donnelly, Proc. R. Soc. B, 2015, 282, 20151684 CrossRef PubMed.
- (a) J. R. Hart, Y. Zhang, L. Liao, L. Ueno, L. Du, M. Jonkers, J. R. Yates III and P. K. Vogt, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 1131–1136 CrossRef PubMed; (b) E. R. Cantwell-Dorris, J. J. O'Leary and O. M. Sheils, Mol. Cancer Ther., 2011, 10, 385–394 CrossRef PubMed.
- For a recent discussion on the definitions of SNPs and mutations as single point variants, see: R. Karki, D. Pandya, R. C. Elston and C. Ferlini, BMC Med. Genomics, 2015, 8, 37 CrossRef PubMed.
- For an example of one of the most common commercial assays for SNP genotyping, TaqMan, see: G. Q. Shen, K. G. Abdullah and Q. K. Wang, Methods Mol. Biol., 2009, 578, 293–306 CrossRef PubMed.
- For a recent review of various SNP detection methods largely based on differences in thermodynamic stability of probe-target duplexes, see: K. Knez, D. Spasic, K. P. F. Janssen and J. Lammertyn, Analyst, 2014, 139, 353–370 RSC.
- For a review, see: D. W. Dodd and R. H. E. Hudson, Mini-Rev. Org. Chem., 2009, 6, 378–391 CrossRef.
- (a) A. Okamoto, Y. Saito and I. Saito, J. Photochem. Photobiol., C, 2005, 6, 108–122 CrossRef; (b) N. Venkatesan, Y. J. Seo and B. H. Kim, Chem. Soc. Rev., 2008, 37, 648–663 RSC; (c) Y. J. Guo and D. C. Jamison, BMC Genomics, 2005, 6, 140 CrossRef PubMed; (d) D. J. Hurley, S. E. Seaman, J. C. Mazura and Y. Tor, Org. Lett., 2002, 4, 2305–2308 CrossRef PubMed; (e) G. T. Hwang, Y. J. Seo and B. H. Kim, J. Am. Chem. Soc., 2004, 126, 6528–6529 CrossRef PubMed; (f) P. Cekan and S. T. Sigurdsson, Chem. Commun., 2008, 3393–3395 RSC; (g) K. Tainaka, K. Tanaka, S. Ikeda, K. Nishiza, T. Unzai, Y. Fujiwara, I. Saito and A. Okamoto, J. Am. Chem. Soc., 2007, 129, 4776–4784 CrossRef PubMed; (h) L. Valis, N. Amann and H. A. Wagenknecht, Org. Biomol. Chem., 2005, 3, 36–38 RSC; (i) L. Bethge, D. V. Jarikote and O. Seitz, Bioorg. Med. Chem., 2008, 16, 114–125 CrossRef PubMed; (j) E. Ergen, M. Weber, J. Jacob, A. Herrmann and K. Mullen, Chem. – Eur. J., 2006, 12, 3707–3713 CrossRef PubMed; (k) Y. Saito, A. Suzuki, Y. Okada, Y. Yamasaka, N. Nemoto and I. Saito, Chem. Commun., 2013, 49, 5684–5686 RSC.
- Y. Saito, K. Motegi, S. S. Bag and I. Saito, Bioorg. Med. Chem., 2008, 16, 107–113 CrossRef PubMed.
- Q. Xiao, R. T. Ranasinghe, A. M. P. Tang and T. Brown, Tetrahedron, 2007, 63, 3483–3490 CrossRef.
- S. Berndl, S. D. Dimitrov, F. Menacher, T. Fiebig and H. A. Wagenknecht, Chem. – Eur. J., 2016, 22, 2386–2395 CrossRef PubMed.
- J.-L. H. A. Duprey, G. A. Bullen, Z.-Y. Zhao, D. M. Bassani, A. F. A. Peacock, J. Wilkie and J. H. R. Tucker, ACS Chem. Biol., 2016, 11, 717–721 CrossRef PubMed.
- I. Saito, Y. Saito, K. Hanawa, K. Hayashi, K. Motegi, S. S. Bag, C. Dohno, T. Ichiba, K. Tainaka and A. Okamoto, Pure Appl. Chem., 2006, 78, 2305–2312 Search PubMed.
- E. Socher, D. V. Jarikote, A. Knoll, L. Roglin, J. Burmeister and O. Seitz, Anal. Biochem., 2008, 375, 318–330 CrossRef PubMed.
- J.-L. H. A. Duprey, D. M. Bassani, E. I. Hyde, C. Ludwig, A. Rodger, J. S. Vyle, J. Wilkie, Z.-Y. Zhao and J. H. R. Tucker, Supramol. Chem., 2011, 23, 273–277 CrossRef.
- J.-L. H. A. Duprey, Z.-Y. Zhao, D. M. Bassani, J. Manchester, J. S. Vyle and J. H. R. Tucker, Chem. Commun., 2011, 47, 6629–6631 RSC.
- Z.-Y. Zhao, M. San, J.-L. H. A. Duprey, J. R. Arrand, J. S. Vyle and J. H. R. Tucker, Bioorg. Med. Chem. Lett., 2012, 22, 129–132 CrossRef PubMed.
- N. Moran, D. M. Bassani, J.-P. Desvergne, S. Keiper, P. A. S. Lowden, J. S. Vyle and J. H. R. Tucker, Chem. Commun., 2006, 5003–5005 RSC.
- (a) P. Li, H. He, Z. Wang, M. Feng, H. Jin, Y. Wu, L. Zhang, L. Zhang and X. Tang, Anal. Chem., 2016, 88, 883–889 CrossRef PubMed; (b) M. E. Ostergaard and P. J. Hrdlicka, Chem. Soc. Rev., 2011, 40, 5771–5788 RSC.
- O. Kohler, D. Venkatrao, D. V. Jarikote and O. Seitz, ChemBioChem, 2005, 6, 69–77 CrossRef PubMed.
- J.-L. H. A. Duprey, J. Carr-Smith, S. L. Horswell, J. Kowalski and J. H. R. Tucker, J. Am. Chem. Soc., 2016, 138, 746–749 CrossRef PubMed.
- H. Kashida, X. G. Liang and H. Asanuma, Curr. Org. Chem., 2009, 13, 1065–1084 CrossRef.
- (a) T. Ihara, T. Fujii, M. Mukae, Y. Kitamura and A. Jyo, J. Am. Chem. Soc., 2004, 126, 8880–8881 CrossRef PubMed; (b) M. Mukae, T. Ihara, M. Tabara and A. Jyo, Org. Biomol. Chem., 2009, 7, 1349–1354 RSC; (c) P. Arslan, A. Jyo and T. Ihara, Org. Biomol. Chem., 2010, 8, 4843–4848 RSC.
- Y. Molard, D. M. Bassani, J. P. Desvergne, N. Moran and J. H. R. Tucker, J. Org. Chem., 2006, 71, 8523–8531 CrossRef PubMed.
- C. V. Kumar and E. H. Asuncion, J. Am. Chem. Soc., 1993, 115, 8547–8553 CrossRef.
- M. Manoharan, K. L. Tivel, M. Zhao, K. Nafisi and T. L. Netzel, J. Phys. Chem., 1995, 99, 17461–17472 CrossRef.
- K. Fukui, K. Tanaka, M. Fujitsuka, A. Watanabe and O. Ito, J. Photochem. Photobiol., B, 1999, 50, 18–27 CrossRef.
- D. V. Jarikote, N. Krebs, S. Tannert, B. Roder and O. Seitz, Chem. – Eur. J., 2007, 13, 300–310 CrossRef PubMed.
- (a) A. S. Dvornikov, J. P. Desvergne, D. A. Oulianov, H. Bouas-Laurent and P. M. Rentzepis, Helv. Chim. Acta, 2001, 84, 2520–2532 CrossRef; (b) H. Yao, T. Okada and N. Mataga, J. Phys. Chem., 1989, 93, 7388–7394 CrossRef.
- (a) N. Mataga, H. Yao, T. Okada and W. Rettig, J. Phys. Chem., 1989, 93, 3383–3386 CrossRef; (b) T. Shida, Electronic Absorption Spectra of Radical Ions, Elsevier, Amsterdam, 1988 Search PubMed.
- C. V. Kumar and E. H. Asuncion, J. Chem. Soc., Chem. Commun., 1992, 470–472 RSC.
- H. T. Allawi and J. SantaLucia, Biochemistry, 1998, 37, 9435–9444 CrossRef PubMed.
- A. Rodger, I. S. Blagbrough, G. Adlam and M. L. Carpenter, Biopolymers, 1994, 34, 1583–1593 CrossRef PubMed.
- A. Rodger, S. Taylor, G. Adlam, I. S. Blagbrough and I. S. Haworth, Bioorg. Med. Chem., 1995, 3, 861–872 CrossRef PubMed.
- H. Asanuma, T. Toda, K. Murayama, X. G. Liang and H. Kashida, J. Am. Chem. Soc., 2010, 132, 14702–14703 CrossRef PubMed.
- (a) H. Asanuma, T. Takarada, T. Yoshida, D. Tamaru, X. G. Liang and M. Komiyama, Angew. Chem., Int. Ed., 2001, 40, 2671–2673 CrossRef; (b) H. Asanuma, X. Liang, T. Yoshida and M. Komiyama, ChemBioChem, 2001, 2, 39–44 CrossRef PubMed.
- M. A. Reynolds, T. A. Beck, R. I. Hogrefe, A. Mccaffrey, L. J. Arnold and M. M. Vaghefi, Bioconjugate Chem., 1992, 3, 366–374 CrossRef PubMed.
- (a) Y. Shi, A. Kuzuya, K. Machida and M. Komiyama, Tetrahedron Lett., 2004, 45, 3703–3706 CrossRef; (b) Y. Shi, K. Machida, A. Kuzuya and M. Komiyama, Bioconjugate Chem., 2005, 16, 306–311 CrossRef PubMed; (c) Y. Shi, A. Kuzuya and M. Komiyama, Chem. Lett., 2003, 32, 464–465 CrossRef; (d) K. Fukui, K. Iwane, T. Shimidzu and K. Tanaka, Tetrahedron Lett., 1996, 37, 4983–4986 CrossRef; (e) K. Fukui and K. Tanaka, Nucleic Acids Res., 1996, 24, 3962–3967 CrossRef PubMed; (f) A. Avino, S. Mazzini, R. Ferreira and R. Eritja, Bioorg. Med. Chem., 2010, 18, 7348–7356 CrossRef PubMed.
- J. Manchester, D. M. Bassani, J.-L. H. A. Duprey, L. Giordano, J. S. Vyle, Z.-Y. Zhao and J. H. R. Tucker, J. Am. Chem. Soc., 2012, 134, 10791–10794 CrossRef PubMed.
- (a) V. L. Malinovskii, D. Wenger and R. Haner, Chem. Soc. Rev., 2010, 39, 410–422 RSC; (b) N. Berova, L. Di Bari and G. Pescitelli, Chem. Soc. Rev., 2007, 36, 914–931 RSC.
- N. K. Modukuru, K. J. Snow, B. S. Perrin, J. Thota and C. V. Kumar, J. Phys. Chem. B, 2005, 109, 11810–11818 CrossRef PubMed.
- B. Norden and H. Becker, J. Am. Chem. Soc., 1999, 121, 11947–11952 CrossRef.
- (a) W. B. Tan, A. Bhambhani, M. R. Duff, A. Rodger and C. V. Kumar, Photochem. Photobiol., 2006, 82, 20–30 CrossRef PubMed; (b) R. Lyng, A. Rodger and B. Norden, Biopolymers, 1992, 32, 1201–1214 CrossRef PubMed.
- H. Nishioka, X. G. Liang and H. Asanuma, Chem. – Eur. J., 2010, 16, 2054–2062 CrossRef PubMed.
- A. C. Jones and R. K. Neely, Q. Rev. Biophys., 2015, 48, 244–279 CrossRef PubMed.
- D. Dziuba, P. Jurkiewicz, M. Cebecauer, M. Hof and M. Hocek, Angew. Chem., Int. Ed., 2016, 55, 174–178 CrossRef PubMed.
- C. K. Liang, J. P. Desvergne and D. M. Bassani, Photochem. Photobiol., 2014, 13, 316–323 RSC.
- Y. Nishimura, K. Shimamura, Y. Ohmori, Y. Shinohara and T. Arai, J. Photochem. Photobiol., A, 2011, 218, 69–75 CrossRef.
- E. T. Kool, J. C. Morales and K. M. Guckian, Angew. Chem., Int. Ed., 2000, 39, 990–1009 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ob01710g |
| ‡ In large scale genotyping to identify many variants within a single genome, high throughput sequencing technologies are typically used. |
| § With other read-out methods emerging, the term base discriminating probe (BDP) is suggested as a more convenient descriptor than the hitherto more commonly used base-discriminating fluorophore (BDF). BDFs have in any case often referred only to probes that contain a fluorophore tag directly attached to a nucleoside (e.g. see ref. 6), as opposed to a non-nucleosidic linker, as is the case with this work. |
| This journal is © The Royal Society of Chemistry 2018 |