
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereoselective synthesis of 2,6-trans-4-oxopiperidines using an acid-mediated 6-endo-trig cyclisation†
Jonathan D.
Bell
a,
Alexander H.
Harkiss
a,
Christopher R.
Wellaway
 b and
Andrew
Sutherland
*a
b and
Andrew
Sutherland
*a
aWestCHEM, School of Chemistry, The Joseph Black Building, University of Glasgow, Glasgow G12 8QQ, UK. E-mail: Andrew.Sutherland@glasgow.ac.uk
bAllergic Inflammation Discovery Performance Unit and Respiratory Therapy Area Unit, GlaxoSmithKline Medicines Research Centre, Gunnels Wood Road, Stevenage SG1 2NY, UK
First published on 11th July 2018
Abstract
An acid-mediated 6-endo-trig cyclisation of amine-substituted enones has been developed for the stereoselective synthesis of trans-6-alkyl-2-methyl-4-oxopiperidines. Performed under conditions that prevent removal of the Boc-protecting group or acetal formation, the key cyclisation was found to generate cleanly the 4-oxopiperidine products in high overall yields from a wide range of alkyl substituted enones. The synthetic utility of the trans-6-alkyl-2-methyl-4-oxopiperidines formed from this process was demonstrated with the total synthesis of the quinolizidine alkaloid, (+)-myrtine and the piperidine alkaloid, (−)-solenopsin A.
Introduction
Substituted piperidines are key structural units found as components of a wide range of natural products and pharmaceutically active compounds.1 Within this structural class, compounds bearing a 2,6-trans-dialkylpiperidine core have been isolated from a wide range of sources and found to have important biological and medicinal properties.1 These include (+)-myrtine (1),2 a quinolizidine alkaloid from Vaccinium myrtillus (Ericaceae) and the phenylquinolizidine alkaloid (−)-lasubine I (2),3 isolated from the leaves of Lagerstroemia subcostata (Fig. 1). Natural products bearing just the 2,6-trans-dialkylpiperidine ring include (−)-solenopsin A (3), a component of the venom of the fire ant Solenopsis invicta.4 As well as being an inhibitor of phosphatidylinositol-3-kinase signalling and angiogenesis,5 (−)-solenopsin A (3), has also been shown to display ceramide-like biological activity with the inhibition of the functional Akt (protein kinase B) pathway.6 Another example is (+)-prosopinine (4), a 2,6-trans-dialkylpiperidine alkaloid from Prosopsis africana,7 that has antihypertensive properties and antibiotic activity against Staphyllococcus aureus.8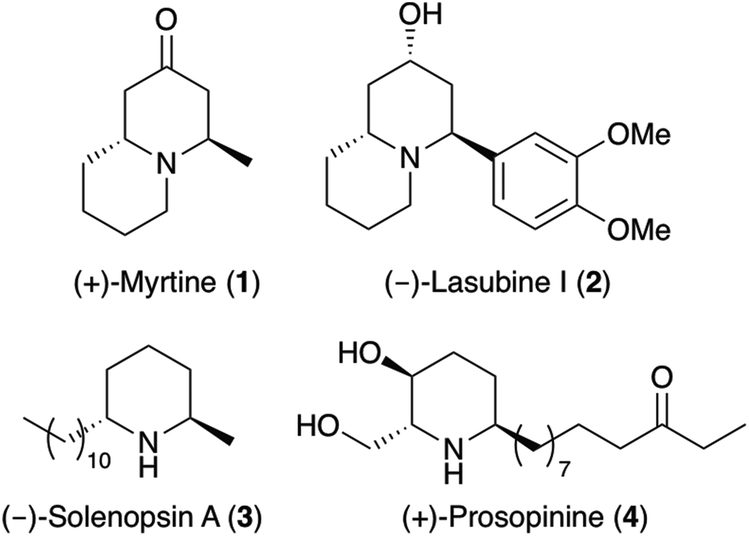 | ||
| Fig. 1 Structures of 2,6-trans-disubstituted piperidine-containing natural products. | ||
As a result of their interesting and varied structures and their significant medicinal properties, a variety of strategies for the total syntheses of 2,6-trans-dialkylpiperidine natural products have been reported.9,10
One approach in accessing the 2,6-trans-dialkylpiperidine ring system is the 6-endo-trig cyclisation11 of amine-substituted enones.12 In 2011, we reported a one-pot, reductive amination/6-endo-trig cyclisation that allowed the synthesis of 2,6-trans-4-oxopipecolic acids.13 A Zimmerman–Traxler chair-like transition state in which both the side-chain and N-substituent occupy a pseudoequatorial position was used to rationalise the stereochemical outcome of the kinetically-controlled cyclisation (Scheme 1a). Troin and co-workers also described the general synthesis of 2,6-trans-dialkylpiperidines by the intramolecular Michael-type reaction of carbamate-protected amine-substituted enones using a combination of p-tosic acid, ethylene glycol and trimethyl orthoformate (Scheme 1b).14 This produced the ketal derivatives of the 2,6-trans-dialkyl-4-oxopiperidines that were isolated by conversion to the more stable thioketals. Recently, we have been reinvestigating the factors that control the stereochemical outcome of the 6-endo-trig cyclisation of amine-substituted enones for the synthesis of piperidine alkaloids.15,16 Herein, we now report the optimisation and scope of a novel acid-mediated 6-endo-trig cyclisation for the preparation of a series of 2,6-trans-2-methyl-4-oxo-6-alkylpiperidines (Scheme 1c). We also describe the use of this chemistry for the total synthesis of two piperidine alkaloids, (+)-myrtine (1) and (−)-solenopsin A (3).
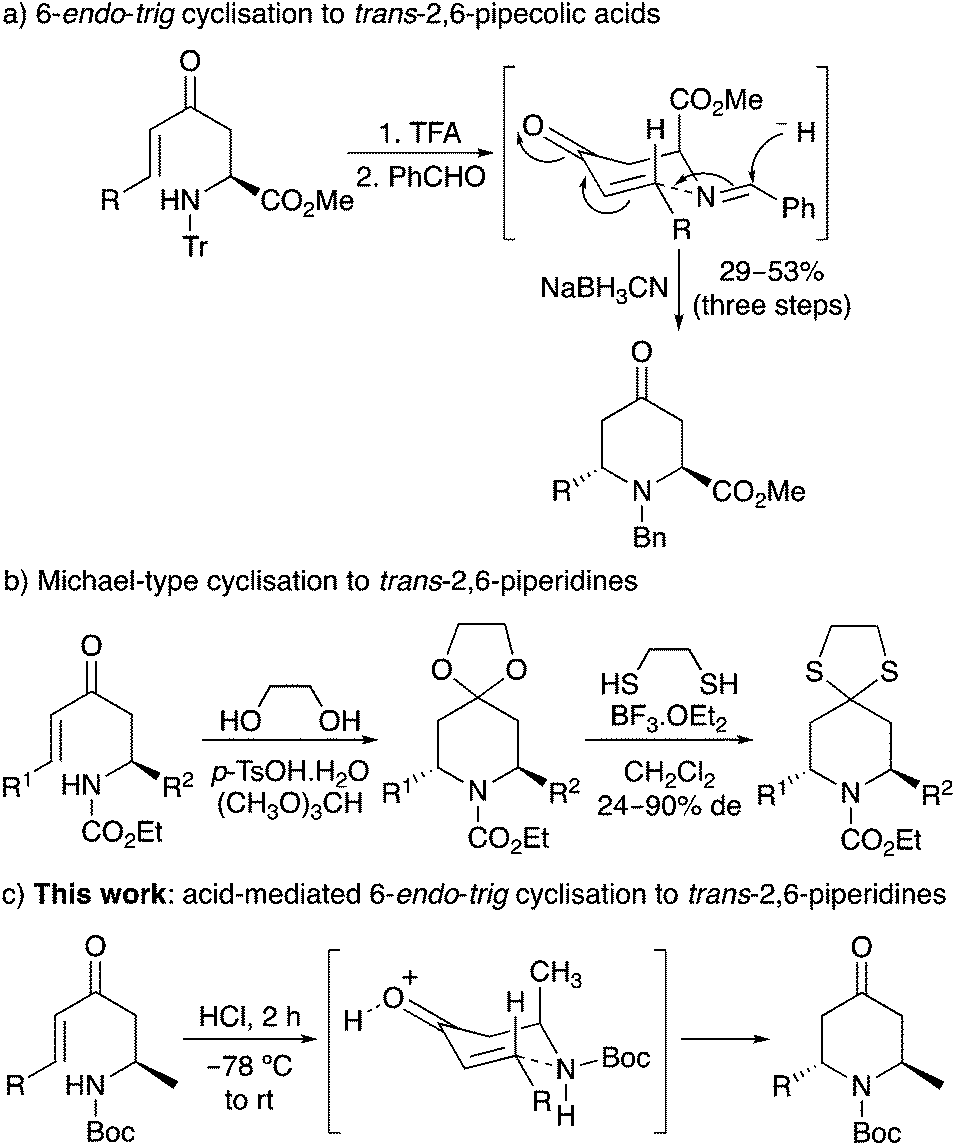 | ||
| Scheme 1 Methods for the synthesis of trans-2,6-disubstituted piperidines. | ||
Results and discussion
This study began with the synthesis of a range of amine-substituted enones. A highly efficient and scalable four-step route from commercially available N-Boc-L-aspartic acid 4-methyl ester (5) was used to access key β-ketophosphonate ester 9 (Scheme 2).16 Chemoselective reduction of 5via the N-hydroxysuccinimide ester using sodium borohydride was followed by a one-step iodination of the resulting alcohol 6 using triphenylphosphine, imidazole and iodine.17,18 Hydrodehalogenation under basic conditions19 gave β-homoalanine derivative 8. Subsequent reaction with the lithium anion of dimethyl methylphosphonate completed the scalable, four-step synthesis of β-ketophosphonate ester 9 in 72% overall yield.20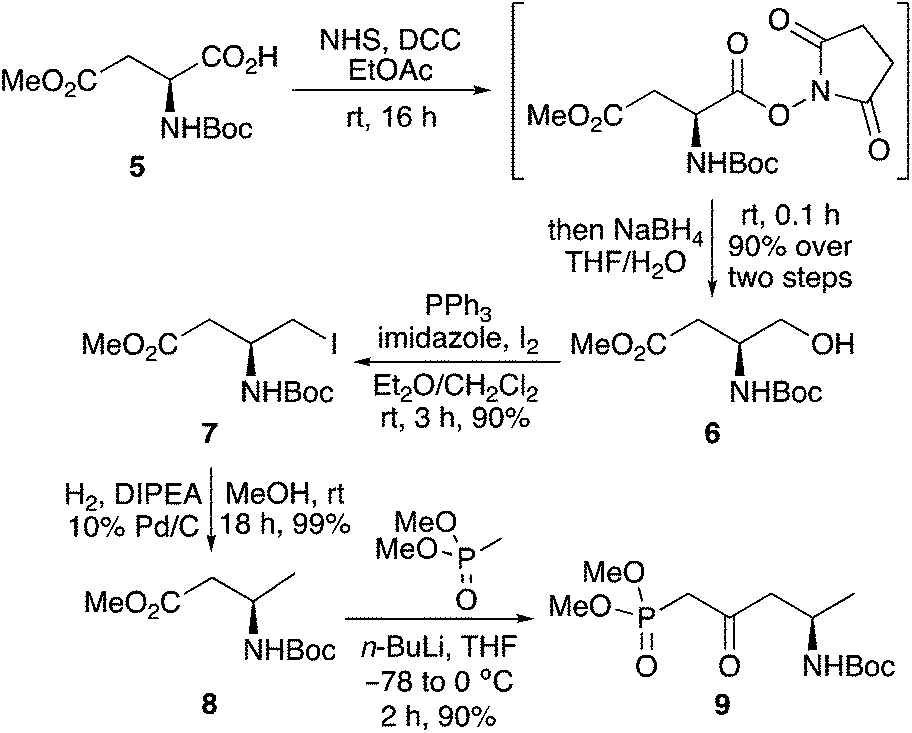 | ||
| Scheme 2 Synthesis of β-ketophosphonate ester 9. | ||
A small library of amine-substituted enones were prepared via the highly stereoselective Horner–Wadsworth–Emmons reaction of 9 with various alkyl and aryl aldehydes, using potassium carbonate as a base, under mild conditions (Scheme 3). This allowed the isolation of E-enones 10a–10l as the sole products in 70–97% yield.16
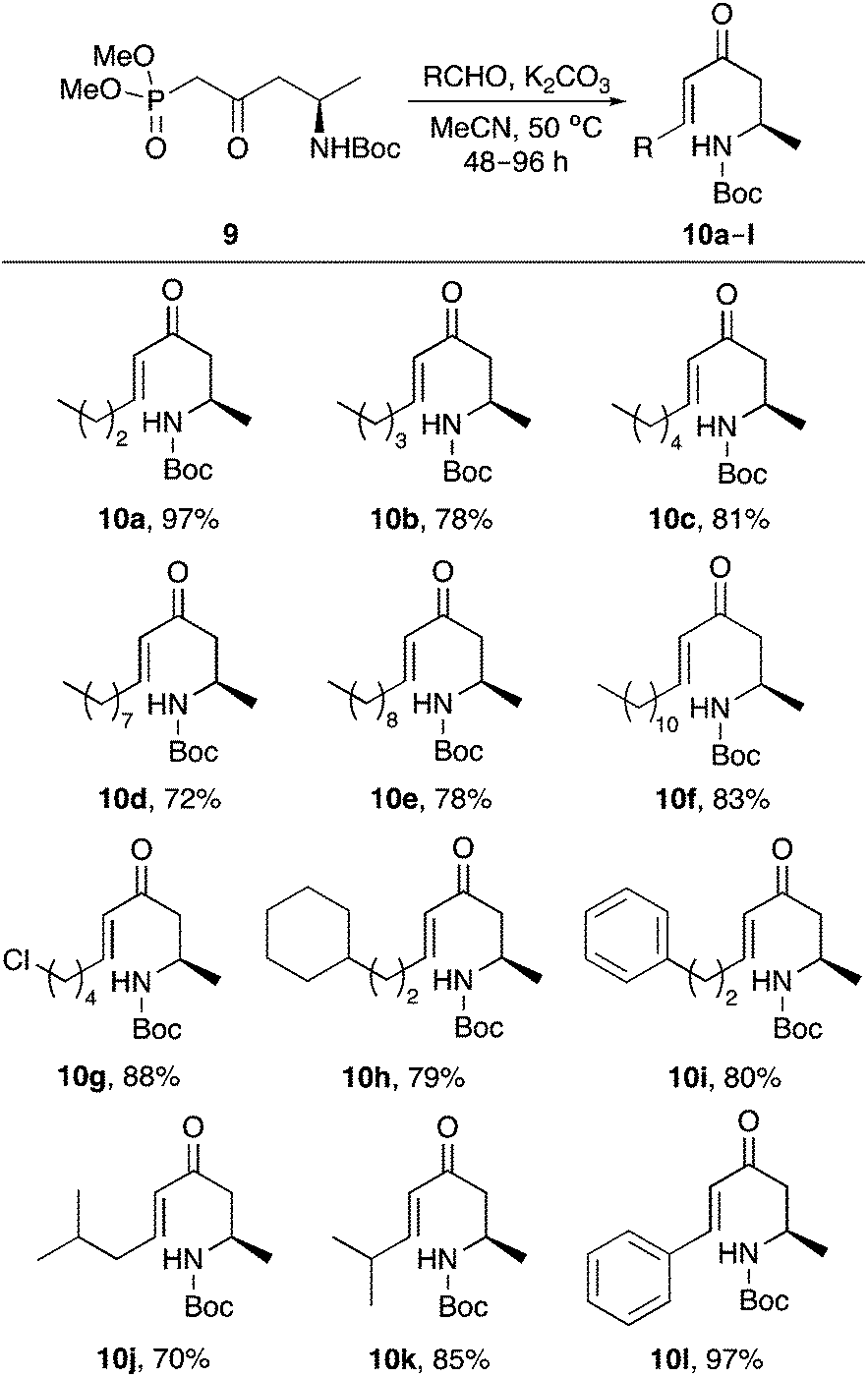 | ||
| Scheme 3 Synthesis of E-enones 10a–l. | ||
Enone 10a was then treated with an ethereal solution of 2 M hydrochloric acid in methanol at room temperature (20 °C). After 1 h, TLC showed the formation of two new compounds. Characterisation by 1H NMR spectroscopy identified these as the 2,6-trans- and 2,6-cis-4-oxopiperidines (11a and 12a), formed in a combined 58% yield and a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, respectively (Table 1, entry 1). Despite the acidic conditions, only trace amounts (<5%) of the deprotected amine or the oxopiperidine derived ketals could be detected by NMR spectroscopy. To explore the limits of the acid mediated 6-endo-trig cyclisation, the reaction was repeated with varying amounts of acid. Using an excess (5 equiv.), quickly led to decomposition of the starting material and the isolation of no major products (entry 2), while using 50 mol% resulted in an incomplete reaction even after 3 h and isolation of 11a and 12a in only 40% yield (entry 3). The temperature of the transformation was then investigated in attempt to improve both the diastereoselectivity and the overall yield. Although cooling the reaction to either 0 or −78 °C and warming to room temperature over 2 h did result in cleaner transformations and significantly improved yields (entries 4 and 5), both reactions again gave 11a and 12a in a 3:1 ratio, respectively. Nevertheless, with the improved efficiency of the cyclisation, and the straightforward separation of the two cyclic products by column chromatography, this allowed the isolation of the major diastereomer 11a in 49% yield.
:1 ratio, respectively (Table 1, entry 1). Despite the acidic conditions, only trace amounts (<5%) of the deprotected amine or the oxopiperidine derived ketals could be detected by NMR spectroscopy. To explore the limits of the acid mediated 6-endo-trig cyclisation, the reaction was repeated with varying amounts of acid. Using an excess (5 equiv.), quickly led to decomposition of the starting material and the isolation of no major products (entry 2), while using 50 mol% resulted in an incomplete reaction even after 3 h and isolation of 11a and 12a in only 40% yield (entry 3). The temperature of the transformation was then investigated in attempt to improve both the diastereoselectivity and the overall yield. Although cooling the reaction to either 0 or −78 °C and warming to room temperature over 2 h did result in cleaner transformations and significantly improved yields (entries 4 and 5), both reactions again gave 11a and 12a in a 3:1 ratio, respectively. Nevertheless, with the improved efficiency of the cyclisation, and the straightforward separation of the two cyclic products by column chromatography, this allowed the isolation of the major diastereomer 11a in 49% yield.
a
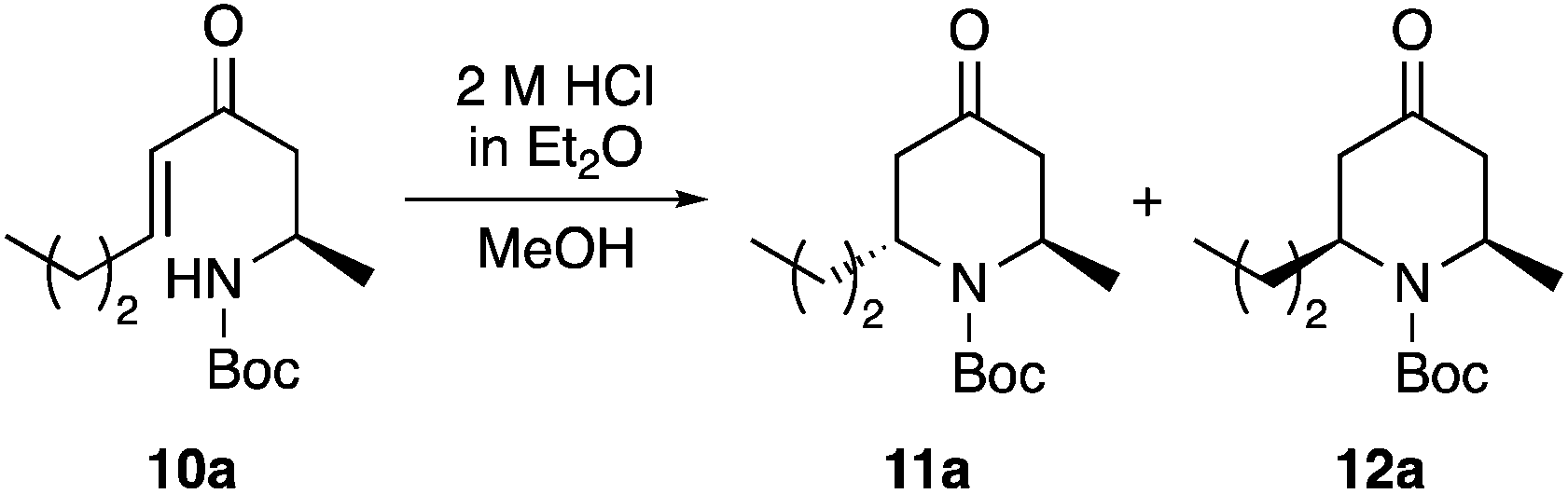
To provide some insight into the mechanism of the acid-mediated 6-endo-trig cyclisation, some control experiments were performed. Initially, the cyclisation of enone 10a over a 24 h period (Fig. 2) was analysed by 1H NMR spectroscopy. As observed in the preparative reaction, the transformation was complete after 2 h, giving 2,6-trans-11a and 2,6-cis-12a in a 76:24 ratio. After 2 h, a steady decrease in the amount of 11a and a concurrent increase in 12a was observed, with a final ratio of 67:33 in favour of 2,6-cis-product 12a (see ESI† for 1H NMR spectra for 2–24 h). Extending the reaction time to 40 h, showed no appreciable change in ratio (69:31). An additional experiment was performed involving resubmission of diastereomerically pure 2,6-trans-product 11a to the acid-mediated cyclisation conditions. Over a 6 h period, 1H NMR analysis of the reaction mixture showed increasing epimerisation of 11a with a final 68:32 ratio of 11a and 12a, respectively. These results taken together suggest that the 2,6-trans-4-oxopiperidine is the less stable, kinetic diastereomer and thus, is formed first during the thermodynamically controlled 6-endo-trig cyclisation. On extension of the reaction time and in the presence of acid, a likely retro-conjugate addition reaction results in conversion of the 2,6-trans-4-oxopiperidine to the configurationally more stable 2,6-cis-isomer.21,22 Therefore, to maximise selective formation of the target 2,6-trans-isomers, short reaction times should be used. While a reaction time of less than 2 h would likely lead to a more selective cyclisation, the optimisation studies have shown that a 2 h reaction time is required for full conversion. Therefore, in terms of maximising the isolation of the target 2,6-trans-isomers, a 2 h reaction is a good compromise.
 | ||
| Fig. 2 Diastereomeric ratio of 11a and 12a during an extended 6-endo-trig cyclisation reaction. | ||
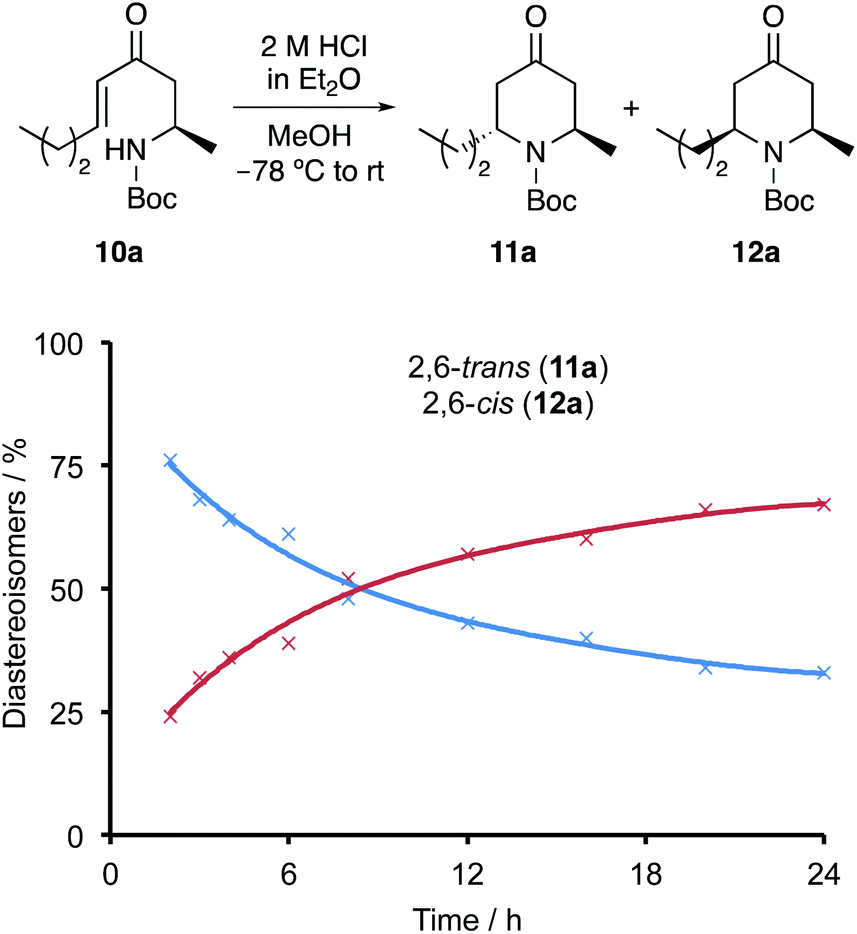
Following optimisation of the acid-mediated cyclisation and with some understanding of the mechanism, the scope of the process was next investigated (Scheme 4). The 6-endo-trig cyclisation of a range of enones 10a–10f bearing n-alkyl side-chains typically present in piperidine alkaloid natural products were found to be excellent substrates for this transformation, giving a mixture of the 2,6-trans- and 2,6-cis-4-oxopiperidines (11 and 12) in high yields (73–88%). Inspection of the 1H NMR spectra from the crude reaction mixtures showed that the 2,6-trans-4-oxopiperidine 11 was the major product in each case with a diastereomeric ratio of approximately 2.5:1 to 3:1. Formation of 2,6-trans-4-oxopiperidines 11a–11f as the major diastereomers during the 2 h acid mediated 6-endo-trig cyclisation and in similar ratios irrespective of the size of the n-alkyl side-chain provides further evidence that these are the kinetic products from this process. For each case, the 2,6-trans-4-oxopiperidines 11a–11f were separated by column chromatography, allowing the isolation of these compounds in 47–55% yields. Further examination of the scope of the cyclisation with enones bearing functional groups (10g) and incorporating rings (10h and 10i), gave the corresponding 2,6-trans-4-oxopiperidines (11g–11i) in similar diastereomeric ratios and isolated yields. Limitations of the cyclisation were observed. For example, enones 10j and 10k with a branched side-chain adjacent to the alkene moiety showed no diastereoselectivity. While the cyclisation of 10j with a sec-butyl side-chain still resulted in an efficient cyclisation and the isolation of each diastereomer in 40% yield (80% overall), the cyclisation of the iso-propyl analogue 10k was impeded, generating the 2,6-trans- and 2,6-cis-4-oxopiperidines 11k/12k in only 54% yield. The other limitation discovered during this study is that enones with a conjugated aryl side-chain (e.g.10l) are not cyclised under the mild conditions of this transformation.
 | ||
| Scheme 4 Scope of the acid-mediated cyclisation for the preparation of 2,6-dialkyl-4-oxopiperidines 11 and 12. | ||
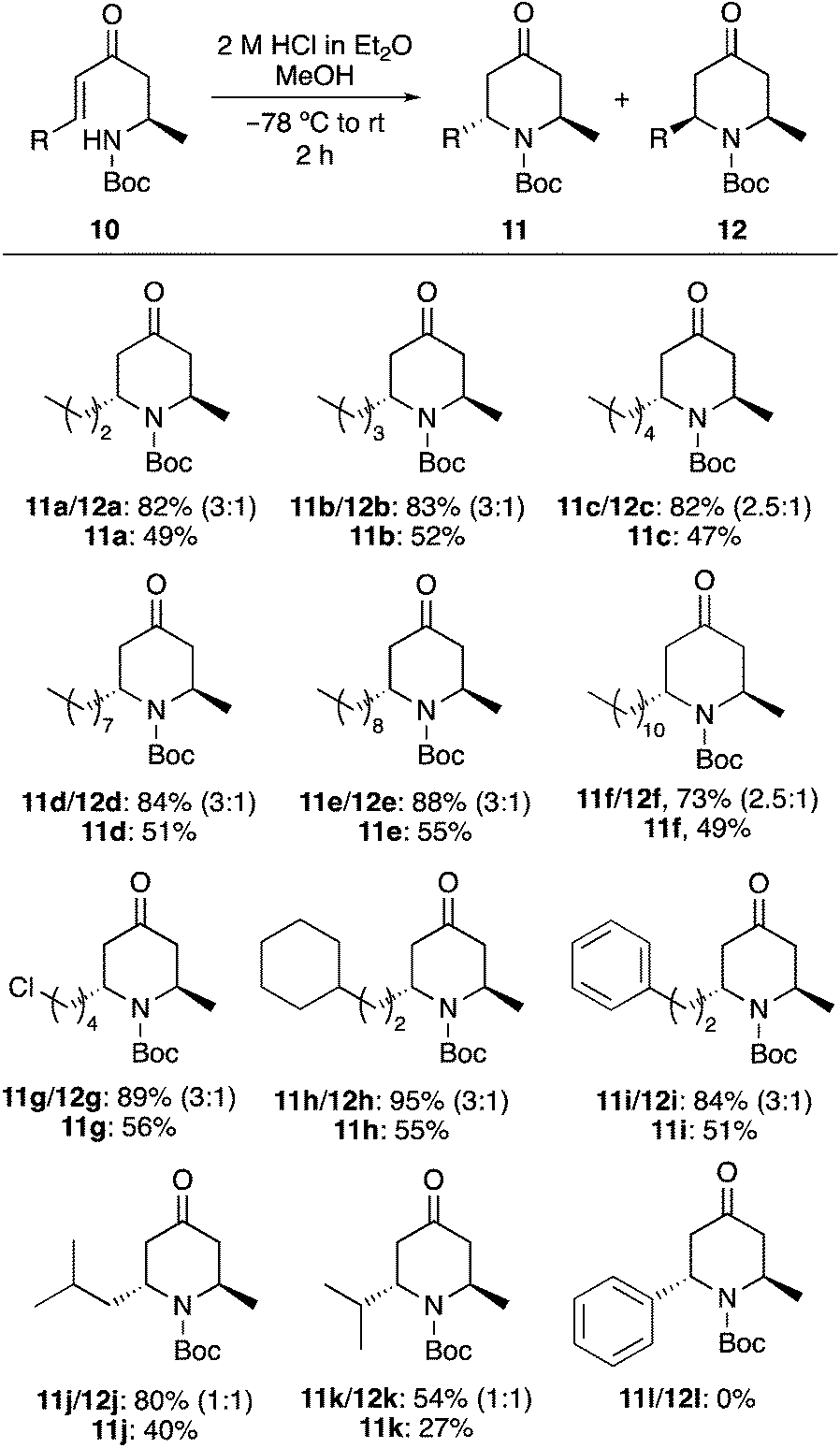
Having developed a new approach for the synthesis of 2,6-trans-dialkyl-4-oxopiperidines, the synthetic utility of this transformation for the facile preparation of piperidine based natural products was next investigated (Scheme 5). Initially, 2,6-trans-4-oxopiperidine 11g bearing a 4-chlorobutyl side-chain was converted in two steps to the quinolizidine alkaloid, (+)-myrtine (1). While the N-Boc protecting group of 11g was stable under the anhydrous acidic conditions of the cyclisation reaction, facile removal was achieved under aqueous acidic conditions. This was followed by cyclisation of the resulting amine with the 4-chlorobutyl side-chain under basic conditions, which gave (+)-myrtine (1) in 62% yield over the two steps. 2,6-trans-4-Oxopiperidine 11f was used for a two-step synthesis of the piperidine alkaloid, (−)-solenopsin A (3). Treatment of 11f with 1,3-propanedithiol under Lewis acid conditions resulted in simultaneous formation of the cyclic dithioketal and removal of the Boc-protecting group. This gave 1,5-dithia-9-azaspiro[5.5]undecane 13 in 90% yield. Reduction of the dithioketal using RANEY® nickel then gave (−)-solenopsin A (3) in 77% yield. The spectroscopic data and optical rotations of 1 and 3 generated in this study were entirely consistent with literature data.10b,14b
 | ||
| Scheme 5 Synthesis of (+)-myrtine (1) and (−)-solenopsin A (3). | ||
Conclusions
In summary, an acid-mediated 6-endo-trig cyclisation of amine-substituted enones has been optimised for the stereoselective synthesis of 2,6-trans-6-alkyl-2-methyl-4-oxopiperidines. Mechanistic studies have shown that the 2,6-trans-4-oxopiperidine is the kinetic product, that is then converted over time via a possible retro-conjugate addition reaction to the configurationally more stable 2,6-cis-isomer. Therefore, using a short reaction time, the scope of the process was explored with a range of amine-substituted enones bearing alkyl side-chains, resulting in the preparation of a small library of 2,6-trans-6-alkyl-2-methyl-4-oxopiperidines. Despite the acidic conditions, neither Boc-group removal or acetal formation was observed, with the Boc-protected 4-oxopiperidines isolated as the major products in 54–95% yield. The potential of the 2,6-trans-4-oxopiperidines as building blocks was demonstrated with the two-step preparation of the quinolizidine alkaloid, (+)-myrtine (1) and the piperidine alkaloid, (−)-solenopsin A (3). Work is currently underway to investigate further synthetic applications of this acid-mediated 6-endo-trig cyclisation reaction.Experimental
All reagents and starting materials were obtained from commercial sources and used as received. All dry solvents were purified using a solvent purification system. All reactions were performed in oven-dried glassware under an atmosphere of argon unless otherwise stated. Brine refers to a saturated solution of sodium chloride. Flash column chromatography was performed using silica gel 60 (40–63 μm). Aluminium-backed plates pre-coated with silica gel 60F254 were used for thin layer chromatography and were visualised with a UV lamp or by staining with potassium permanganate. 1H NMR spectra were recorded on a NMR spectrometer at either 400 or 500 MHz and data are reported as follows: chemical shift in ppm relative to tetramethylsilane or the solvent as the internal standard (CDCl3, δ 7.26 ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet or overlap of nonequivalent resonances, integration). 13C NMR spectra were recorded on a NMR spectrometer at either 101 or 126 MHz and data are reported as follows: chemical shift in ppm relative to tetramethylsilane or the solvent as internal standard (CDCl3, δ 77.0 ppm), multiplicity with respect to hydrogen (deduced from DEPT experiments, C, CH, CH2 or CH3). IR spectra were recorded on a FTIR spectrometer; wavenumbers are indicated in cm−1. Mass spectra were recorded using electrospray or electron impact techniques. HRMS spectra were recorded using a dual-focusing magnetic analyser mass spectrometer. Melting points are uncorrected. Optical rotations were determined as solutions irradiating with the sodium D line (λ = 589 nm) using a polarimeter. [α]D values are given in units 10−1 deg cm2 g−1.Methyl (3S)-3-(tert-butoxycarbonylamino)-4-hydroxybutanoate (6)23
To a solution of N-Boc-L-aspartic acid 4-methyl ester (5) (8.27 g, 33.5 mmol) in ethyl acetate (200 mL) at 0 °C was added N-hydroxysuccinimide (4.24 g, 36.9 mmol). N,N-Dicyclohexylcarbodiimide (7.05 g, 34.2 mmol) in ethyl acetate (20 mL) was then added dropwise. The reaction mixture was allowed to warm to room temperature and stirred for 16 h. Once the reaction was complete, the reaction mixture was filtered through Celite. The filtrate was washed with saturated sodium carbonate solution (100 mL), brine (100 mL), dried (MgSO4) and concentrated in vacuo. The resulting residue was then dissolved in tetrahydrofuran (20 mL) and added dropwise to a solution of sodium borohydride (2.03 g, 53.6 mmol) in a mixture of tetrahydrofuran and water (7.5:1, 85 mL). The reaction mixture was stirred for 0.1 h before quenching with saturated aqueous ammonium chloride (5 mL). The reaction mixture was extracted with dichloromethane (3 × 50 mL). The organic fractions were combined and washed with brine (100 mL), dried (MgSO4), filtered and concentrated in vacuo. Purification by flash column chromatography using silica gel, eluting with 30% ethyl acetate in dichloromethane gave methyl (3S)-3-(tert-butoxycarbonylamino)-4-hydroxybutanoate (6) as a colourless oil (7.03 g, 90%). [α]26D +5.6 (c 1.0, CHCl3), lit.23 [α]23D +6.3 (c 0.5, CHCl3); δH (400 MHz, CDCl3) 1.42 (9H, s, 3 × CH3), 2.62 (2H, d, J 6.1 Hz, 2-H2), 2.82 (1H, br s, OH), 3.65–3.73 (5H, m, OCH3 and 4-H2), 3.92–4.04 (1H, m, 3-H), 5.25 (1H, br s, NH); δC (126 MHz, CDCl3) 28.4 (3 × CH3), 35.9 (CH2), 49.4 (CH), 52.0 (CH3), 64.4 (CH2), 79.9 (C), 155.9 (C), 172.4 (C); m/z (ESI) 256 (MNa+, 100%).
Methyl (3S)-3-(tert-butoxycarbonylamino)-4-iodobutanoate (7)17
To a suspension of imidazole (4.11 g, 60.4 mmol) and triphenylphosphine (11.9 g, 45.3 mmol) in a mixture of diethyl ether and dichloromethane (2:1, 100 mL) at 0 °C was added iodine (11.5 g, 45.3 mmol) in three portions over 0.5 h. After stirring for a further 0.2 h, a solution of methyl (3S)-3-(tert-butoxycarbonylamino)-4-hydroxybutanoate (6) (7.03 g, 30.2 mmol) in a mixture of diethyl ether and dichloromethane (2:1, 50 mL) was added and the resulting mixture was stirred for 3 h at room temperature. The reaction mixture was filtered through Celite and the filtrate was concentrated in vacuo. Purification by flash column chromatography using silica gel, eluting with 30% diethyl ether in petroleum ether (40–60) gave methyl (3S)-3-(tert-butoxycarbonylamino)-4-iodobutanoate (7) (9.33 g, 90%) as a colourless oil. Spectroscopic data were consistent with the literature.17 [α]33D +7.3 (c 1.0, CHCl3); δH (400 MHz, CDCl3) 1.40 (9H, s, 3 × CH3), 2.60 (1H, dd, J 16.4, 6.1 Hz, 2-HH), 2.70 (1H, dd, J 16.4, 5.6 Hz, 2-HH), 3.28–3.45 (2H, m, 4-H2), 3.66 (3H, s, OCH3), 3.81–3.95 (1H, m, 3-H), 5.11 (1H, d, J 7.2 Hz, NH); δC (101 MHz, CDCl3) 11.1 (CH2), 28.5 (3 × CH3), 38.5 (CH2), 47.7 (CH), 52.0 (CH3), 79.9 (C), 154.7 (C), 171.1 (C); m/z (ESI) 366 (MNa+, 100%).
Methyl (3R)-3-(tert-butoxycarbonylamino)butanoate (8)24
A solution of methyl (3S)-3-(tert-butoxycarbonylamino)-4-iodobutanoate (7) (9.33 g, 27.2 mmol), N,N-diisopropylethylamine (7.11 mL, 40.8 mmol) and 10% Pd/C (2.89 g) in methanol (50 mL) were purged with hydrogen for 0.5 h. The reaction mixture was stirred under an atmosphere of hydrogen for 18 h at room temperature. The mixture was then filtered through Celite and the filtrate was concentrated in vacuo. The resulting residue was dissolved in dichloromethane (100 mL) and washed with a saturated solution of sodium hydrogen carbonate (50 mL), 1 M hydrochloric acid (50 mL), brine (50 mL), dried over MgSO4, filtered and concentrated in vacuo to give methyl (3R)-3-(tert-butoxycarbonylamino)butanoate (8) as a colourless oil (5.87 g, 99%). Spectroscopic data were consistent with the literature.24 [α]26D +21.5 (c 1.0, CHCl3); δH (500 MHz, CDCl3) 1.20 (3H, d, J 6.8 Hz, 4-H3), 1.43 (9H, s, 3 × CH3), 2.47 (1H, dd, J 15.5, 6.0 Hz, 2-HH), 2.52 (1H, dd, J 15.5, 5.4 Hz, 2-HH), 3.68 (3H, s, OCH3), 4.03 (1H, br s, 3-H), 4.91 (1H, br s, NH); δC (126 MHz, CDCl3) 20.5 (CH3), 28.4 (3 × CH3), 40.7 (CH2), 43.5 (CH), 51.6 (CH3), 79.3 (C), 155.1 (C), 172.0 (C); m/z (ESI) 240 (MNa+, 100%).(4R)-4-(tert-Butoxycarbonylamino)-1-(dimethyloxyphosphoryl)pentan-2-one (9)
Dimethyl methylphosphonate (3.74 mL, 34.5 mmol) was dissolved in tetrahydrofuran (100 mL) and cooled to −78 °C under an argon atmosphere. n-Butyl lithium (2.5 M, in hexane, 13.8 mL, 34.5 mmol) was added dropwise and the mixture was stirred for 0.5 h. A solution of methyl (3R)-3-(tert-butoxycarbonylamino)butanoate (8) (3.00 g, 13.8 mmol) in tetrahydrofuran (20 mL) was added dropwise. The resulting mixture was then stirred at −78 °C for 0.5 h and allowed to warm to 0 °C over a period of 1 h. The reaction was quenched with a saturated aqueous solution of ammonium chloride (4 mL) and extracted with ethyl acetate (2 × 50 mL). The combined organic layers were combined, washed with brine (100 mL), dried (MgSO4), filtered and concentrated in vacuo. Purification by flash column chromatography using silica gel, eluting with 40% ethyl acetate in dichloromethane gave (4R)-4-(tert-butoxycarbonylamino)-1-(dimethyloxyphosphoryl)pentan-2-one (9) (3.84 g, 90%) as a colourless oil. νmax/cm−1 (neat) 3316 (NH), 2976 (CH), 1704 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1700 (CO), 1248, 1167, 1023; [α]31D +38.3 (c 1.0, CHCl3); δH (400 MHz, CDCl3) 1.17 (3H, d, J 6.7 Hz, 5-H3), 1.39 (9H, s, 3 × CH3), 2.71 (1H, dd, J 16.9, 5.9 Hz, 3-HH), 2.80 (1H, dd, J 16.9, 5.9 Hz, 3-HH), 3.03 (1H, dd, J 22.6, 13.6 Hz, 1-HH), 3.12 (1H, dd, J 22.6, 13.6 Hz, 1-HH), 3.74 (3H, d, J 0.8 Hz, OCH3), 3.77 (3H, d, J 0.8 Hz, OCH3), 3.93–4.09 (1H, m, 4-H), 4.92 (1H, br s, NH); δC (101 MHz, CDCl3) 20.5 (CH3), 28.3 (3 × CH3), 41.8 (d, JC–P 127.5 Hz, CH2), 43.2 (CH), 50.1 (CH2), 53.0 (d, JC–O–P 6.5 Hz, CH3), 53.1 (d, JC–O–P 6.5 Hz, CH3), 79.2 (C), 155.1 (C), 200.5 (C); m/z (ESI) 332.1225 (MNa+. C12H24NNaO6P requires 332.1233).
O), 1700 (CO), 1248, 1167, 1023; [α]31D +38.3 (c 1.0, CHCl3); δH (400 MHz, CDCl3) 1.17 (3H, d, J 6.7 Hz, 5-H3), 1.39 (9H, s, 3 × CH3), 2.71 (1H, dd, J 16.9, 5.9 Hz, 3-HH), 2.80 (1H, dd, J 16.9, 5.9 Hz, 3-HH), 3.03 (1H, dd, J 22.6, 13.6 Hz, 1-HH), 3.12 (1H, dd, J 22.6, 13.6 Hz, 1-HH), 3.74 (3H, d, J 0.8 Hz, OCH3), 3.77 (3H, d, J 0.8 Hz, OCH3), 3.93–4.09 (1H, m, 4-H), 4.92 (1H, br s, NH); δC (101 MHz, CDCl3) 20.5 (CH3), 28.3 (3 × CH3), 41.8 (d, JC–P 127.5 Hz, CH2), 43.2 (CH), 50.1 (CH2), 53.0 (d, JC–O–P 6.5 Hz, CH3), 53.1 (d, JC–O–P 6.5 Hz, CH3), 79.2 (C), 155.1 (C), 200.5 (C); m/z (ESI) 332.1225 (MNa+. C12H24NNaO6P requires 332.1233).
(2R,5E)-2-(tert-Butoxycarbonylamino)-4-oxonona-5-ene (10a)
(4R)-4-(tert-Butoxycarbonylamino)-1-(dimethyloxyphosphoryl)pentan-2-one (9) (0.421 g, 1.36 mmol) was dissolved in anhydrous acetonitrile (14 mL) and potassium carbonate (0.225 g, 1.63 mmol) was added. The mixture was stirred at room temperature for 0.5 h followed by addition of butyraldehyde (0.250 mL, 2.72 mmol). The temperature was increased to 50 °C and the mixture stirred for 72 h. The solution was then concentrated in vacuo, redissolved in ethyl acetate (20 mL) and washed with water (2 × 15 mL) and then brine (15 mL). The organic layer was dried (MgSO4) and concentrated in vacuo. Purification using a plug of silica gel, eluting with 20% ethyl acetate in petroleum ether (40–60) gave (2R,5E)-2-(tert-butoxycarbonylamino)-4-oxonona-5-ene (10a) as a clear colourless oil (0.375 g, 97%). νmax/cm−1 (neat) 3350 (NH), 2968 (CH), 1689 (CO), 1516, 1365, 1166, 1053, 977; [α]26D +9.3 (c 1.0, CHCl3); δH (500 MHz, CDCl3) 0.94 (3H, t, J 7.3 Hz, 9-H3), 1.20 (3H, d, J 6.9 Hz, 1-H3), 1.43 (9H, s, 3 × CH3), 1.46–1.55 (2H, m, 8-H2), 2.20 (2H, qd, J 6.9, 1.5 Hz, 7-H2), 2.64 (1H, dd, J 15.9, 6.5 Hz, 3-HH), 2.86 (1H, dd, J 15.9, 4.5 Hz, 3-HH), 3.98–4.09 (1H, m, 2-H), 5.02 (1H, br s, NH), 6.09 (1H, dt, J 15.9, 1.5 Hz, 5-H), 6.86 (1H, dt, J 15.9, 6.9 Hz, 6-H); δC (126 MHz, CDCl3) 13.7 (CH3), 20.5 (CH3), 21.3 (CH2), 28.4 (3 × CH3), 34.5 (CH2), 43.7 (CH), 45.7 (CH2), 79.1 (C), 130.8 (CH), 148.2 (CH), 155.2 (C), 199.2 (C); m/z (ESI) 278.1725 (MNa+. C14H25NNaO3 requires 278.1727).
(2R,5E)-2-(tert-Butoxycarbonylamino)-4-oxodec-5-ene (10b)
The reaction was carried out according to the above procedure for the synthesis of (2R,5E)-2-(tert-butoxycarbonylamino)-4-oxonona-5-ene (10a) using (4R)-4-(tert-butoxycarbonylamino)-1-(dimethyloxyphosphoryl)pentan-2-one (9) (0.100 g, 0.326 mmol) and valeraldehyde (0.070 mL, 0.652 mmol). Purification by flash column chromatography using silica gel, eluting with 20% diethyl ether in petroleum ether (40–60) gave (2R,5E)-2-(tert-butoxycarbonylamino)-4-oxodec-5-ene (10b) (0.069 g, 78%) as a colourless oil. νmax/cm−1 (neat) 3356 (NH), 2961 (CH), 1693 (CO), 1516, 1366, 1248, 1171, 1053; [α]20D +6.1 (c 0.9, CHCl3); δH (400 MHz, CDCl3) 0.88 (3H, t, J 7.2 Hz, 10-H3), 1.17 (3H, d, J 6.7 Hz, 1-H3), 1.27–1.46 (13H, m, 3 × CH3, 8-H2, 9-H2), 2.19 (2H, qd, J 7.1, 1.5 Hz, 7-H2), 2.60 (1H, dd, J 15.9, 6.6 Hz, 3-HH), 2.82 (1H, dd, J 15.9, 4.9 Hz, 3-HH), 3.94–4.07 (1H, m, 2-H), 4.95 (1H, br s, NH), 6.06 (1H, dt, J 15.9, 1.5 Hz, 5-H), 6.82 (1H, dt, J 15.9, 7.1 Hz, 6-H); δC (101 MHz, CDCl3) 13.7 (CH3), 20.5 (CH3), 22.2 (CH2), 28.4 (3 × CH3), 30.1 (CH2), 32.1 (CH2), 43.7 (CH), 45.7 (CH2), 79.1 (C), 130.6 (CH), 148.4 (CH), 155.1 (C), 199.0 (C); m/z (ESI) 292.1890 (MNa+. C15H27NNaO3 requires 292.1889).
(2R,5E)-2-(tert-Butoxycarbonylamino)-4-oxoundec-5-ene (10c)
The reaction was carried out according to the above procedure for the synthesis of (2R,5E)-2-(tert-butoxycarbonylamino)-4-oxonona-5-ene (10a) using (4R)-4-(tert-butoxycarbonylamino)-1-(dimethyloxyphosphoryl)pentan-2-one (9) (0.047 g, 0.151 mmol) and hexanal (0.037 mL, 0.303 mmol). Purification by flash column chromatography using silica gel, eluting with 20% diethyl ether in petroleum ether (40–60) gave (2R,5E)-2-(tert-butoxycarbonylamino)-4-oxoundec-5-ene (10c) as a colourless oil (0.035 g, 81%). νmax/cm−1 (neat) 3341 (NH), 2928 (CH), 1694 (CO), 1516, 1366, 1173, 1053; [α]24D +5.6 (c 0.7, CHCl3); δH (400 MHz, CDCl3) 0.89 (3H, t, J 6.9 Hz, 11-H3), 1.20 (3H, d, J 6.7 Hz, 1-H3), 1.24–1.36 (4H, m, 9-H2 and 10-H2), 1.40–1.52 (11H, m, 8-H2 and 3 × CH3), 2.14 (2H, qd, J 7.1, 1.4 Hz, 7-H2), 2.63 (1H, dd, J 15.9, 6.5 Hz, 3-HH), 2.85 (1H, dd, J 15.9, 4.6 Hz, 3-HH), 3.95–4.07 (1H, m, 2-H), 4.94 (1H, br s, NH), 6.08 (1H, dt, J 15.8, 1.4 Hz, 5-H), 6.85 (1H, dt, J 15.8, 7.1 Hz, 6-H); δC (101 MHz, CDCl3) 14.1 (CH3), 20.6 (CH3), 22.6 (CH2), 27.9 (CH2), 28.6 (3 × CH3), 31.5 (CH2), 32.6 (CH2), 43.9 (CH), 45.8 (CH2), 79.3 (C), 130.7 (CH), 148.7 (CH), 155.3 (C), 199.3 (C); m/z (ESI) 306.2039 (MNa+. C16H29NNaO3 requires 306.2040).
(2R,5E)-2-(tert-Butoxycarbonylamino)-4-oxotetradec-5-ene (10d)
The reaction was carried out according to the above procedure for the synthesis of (2R,5E)-2-(tert-butoxycarbonylamino)-4-oxonona-5-ene (10a) using (4R)-4-(tert-butoxycarbonylamino)-1-(dimethyloxyphosphoryl)pentan-2-one (9) (0.129 g, 0.416 mmol) and nonanal (0.143 mL, 0.832 mmol). Purification by flash column chromatography using silica gel, eluting with 30% diethyl ether in petroleum ether (40–60) gave (2R,5E)-2-(tert-butoxycarbonylamino)-4-oxotetradec-5-ene (10d) as a colourless oil (0.098 g, 72%). νmax/cm−1 (neat) 3352 (NH), 2924 (CH), 1694 (CO), 1501, 1366, 1246, 1169, 1053; [α]29D +6.5 (c 0.8, CHCl3); δH (400 MHz, CDCl3) 0.87 (3H, t, J 6.9 Hz, 14-H3), 1.19 (3H, d, J 6.7 Hz, 1-H3), 1.22–1.35 (10H, m, 9-H2, 10-H2, 11-H2, 12-H2 and 13-H2), 1.38–1.50 (11H, m, 8-H2 and 3 × CH3), 2.20 (2H, qd, J 7.0, 1.5 Hz, 7-H2), 2.62 (1H, dd, J 15.9, 6.5 Hz, 3-HH), 2.85 (1H, dd, J 15.9, 4.8 Hz, 3-HH), 3.95–4.09 (1H, m, 2-H), 4.95 (1H, br s, NH), 6.07 (1H, dt, J 15.9, 1.5 Hz, 5-H), 6.84 (1H, dt, J 15.9, 7.0 Hz, 6-H); δC (101 MHz, CDCl3) 14.1 (CH3), 20.5 (CH3), 22.7 (CH2), 28.1 (CH2), 28.4 (3 × CH3), 29.2 (2 × CH2), 29.3 (CH2), 31.8 (CH2), 32.5 (CH2), 43.8 (CH), 45.7 (CH2), 79.2 (C), 130.6 (CH), 148.5 (CH), 155.2 (C), 199.1 (C); m/z (ESI) 348.2498 (MNa+. C19H35NNaO3 requires 348.2509).
(2R,5E)-2-(tert-Butoxycarbonylamino)-4-oxopentadec-5-ene (10e)
The reaction was carried out according to the above procedure for the synthesis of (2R,5E)-2-(tert-butoxycarbonylamino)-4-oxonona-5-ene (10a) using (4R)-4-(tert-butoxycarbonylamino)-1-(dimethyloxyphosphoryl)pentan-2-one (9) (0.405 g, 1.31 mmol) and decanal (0.500 mL, 2.62 mmol) for 96 h. Purification by flash column chromatography using silica gel, eluting with 30% diethyl ether in petroleum ether (40–60) gave (2R,5E)-2-(tert-butoxycarbonylamino)-4-oxopentadec-5-ene (10e) (0.345 g, 78%) as a colourless oil. νmax/cm−1 (neat) 3327 (NH), 2958 (CH), 1693 (CO), 1365, 1170, 1053; [α]25D +4.2 (c 1.0, CHCl3); δH (400 MHz, CDCl3) 0.88 (3H, t, J 6.3 Hz, 15-H3), 1.20 (3H, d, J 6.7 Hz, 1-H3), 1.23–1.34 (12H, m, 9-H2, 10-H2, 11-H2, 12-H2, 13-H2 and 14-H2), 1.39–1.50 (11H, m, 8-H2 and 3 × CH3), 2.20 (2H, q, J 7.0 Hz, 7-H2), 2.63 (1H, dd, J 15.7, 6.6 Hz, 3-HH), 2.85 (1H, dd, J 15.7, 4.5 Hz, 3-HH), 3.96–4.08 (1H, m, 2-H), 4.95 (1H, br s, NH), 6.08 (1H, d, J 15.6 Hz, 5-H), 6.85 (1H, dt, J 15.6, 7.0 Hz, 6-H); δC (126 MHz, CDCl3) 14.1 (CH3), 20.5 (CH3), 22.7 (CH2), 28.1 (CH2), 28.4 (3 × CH3), 29.2 (CH2), 29.3 (CH2), 29.4 (CH2), 29.5 (CH2), 31.9 (CH2), 32.5 (CH2), 43.7 (CH), 45.7 (CH2), 79.2 (C), 130.6 (CH), 148.5 (CH), 155.2 (C), 199.2 (C); m/z (ESI) 362.2649 (MNa+. C20H37NNaO3 requires 362.2666).
(2R,5E)-2-(tert-Butoxycarbonylamino)-4-oxoheptadec-5-ene (10f)
The reaction was carried out according to the above procedure for the synthesis of (2R,5E)-2-(tert-butoxycarbonylamino)-4-oxonona-5-ene (10a) using (4R)-4-(tert-butoxycarbonylamino)-1-(dimethyloxyphosphoryl)pentan-2-one (9) (0.750 g, 2.42 mmol) and dodecanal (1.08 mL, 4.85 mmol). Purification by flash column chromatography using silica gel, eluting with 30% diethyl ether in petroleum ether (40–60) gave (2R,5E)-2-(tert-butoxycarbonylamino)-4-oxoheptadec-5-ene (10f) as a colourless oil (0.734 g, 83%). νmax/cm−1 (neat) 3354 (NH), 2925 (CH), 1694 (CO), 1515, 1366, 1248, 1172, 1053; [α]22D +4.8 (c 1.0, CHCl3); δH (400 MHz, CDCl3) 0.87 (3H, t, J 6.8 Hz, 17-H3), 1.19 (3H, d, J 6.8 Hz, 1-H3), 1.22–1.36 (16H, m, 9-H2, 10-H2, 11-H2, 12-H2, 13-H2, 14-H2, 15-H2 and 16-H2), 1.38–1.50 (11H, m, 8-H2, 3 × CH3), 2.20 (2H, qd, J 7.1, 1.4 Hz, 7-H2), 2.63 (1H, dd, J 15.9, 6.6 Hz, 3-HH), 2.85 (1H, dd, J 15.9, 4.7 Hz, 3-HH), 3.93–4.09 (1H, m, 2-H), 4.96 (1H, br s, NH), 6.07 (1H, dt, J 15.8, 1.4 Hz, 5-H), 6.85 (1H, dt, J 15.8, 7.1 Hz, 6-H); δC (101 MHz, CDCl3) 14.3 (CH3), 20.6 (CH3), 22.8 (CH2), 28.2 (CH2), 28.6 (3 × CH3), 29.3 (CH2), 29.5 (2 × CH2), 29.7 (CH2), 29.8 (2 × CH2), 32.0 (CH2), 32.7 (CH2), 43.8 (CH), 45.8 (CH2), 79.3 (C), 130.7 (CH), 148.7 (CH), 155.3 (C), 199.3 (C); m/z (ESI) 390.2985 (MNa+. C22H41NNaO3 requires 390.2979).
(2R,5E)-2-(tert-Butoxycarbonylamino)-10-chloro-4-oxodec-5-ene (10g)
The reaction was carried out according to the above procedure for the synthesis of (2R,5E)-2-(tert-butoxycarbonylamino)-4-oxonona-5-ene (10a) using (4R)-4-(tert-butoxycarbonylamino)-1-(dimethyloxyphosphoryl)pentan-2-one (9) (0.114 g, 0.367 mmol) and 5-chloropentanal (0.089 g, 0.735 mmol) for 96 h. Purification by flash column chromatography using silica gel, eluting with 30% diethyl ether in petroleum ether (40–60) gave (2R,5E)-2-(tert-butoxycarbonylamino)-10-chloro-4-oxodec-5-ene (10g) as a colourless oil (0.098 g, 88%). νmax/cm−1 (neat) 3357 (NH), 2977 (CH), 1693 (CO), 1663 (CO), 1510, 1366, 1247, 1167, 1053; [α]22D +4.7 (c 1.0, CHCl3); δH (400 MHz, CDCl3) 1.19 (3H, d, J 6.8 Hz, 1-H3), 1.42 (9H, s, 3 × CH3), 1.57–1.67 (2H, m, 8-H2), 1.74–1.88 (2H, m, 9-H2), 2.25 (2H, qd, J 6.9, 1.5 Hz, 7-H2), 2.62 (1H, dd, J 15.9, 6.6 Hz, 3-HH), 2.85 (1H, dd, J 15.9, 4.7 Hz, 3-HH), 3.54 (2H, t, J 6.5 Hz, 10-H2), 3.94–4.08 (1H, m, 2-H), 4.94 (1H, br s, NH), 6.09 (1H, dt, J 15.9, 1.5 Hz, 5-H), 6.83 (1H, dt, J 15.9, 6.9 Hz, 6-H); δC (101 MHz, CDCl3) 20.6 (CH3), 25.4 (CH2), 28.5 (3 × CH3), 31.7 (CH2), 32.0 (CH2), 43.8 (CH), 44.7 (CH2), 46.0 (CH2), 79.3 (C), 131.0 (CH), 147.2 (CH), 155.3 (C), 199.0 (C); m/z (ESI) 326.1484 (MNa+. C15H2635ClNNaO3 requires 326.1493).
(2R,5E)-2-(tert-Butoxycarbonylamino)-8-cyclohexyl-4-oxooct-5-ene (10h)
The reaction was carried out according to the above procedure for the synthesis of (2R,5E)-2-(tert-butoxycarbonylamino)-4-oxonona-5-ene (10a) using (4R)-4-(tert-butoxycarbonylamino)-1-(dimethyloxyphosphoryl)pentan-2-one (9) (0.044 g, 0.144 mmol) and 3-cyclohexylpropanal (0.040 g, 0.288 mmol). Purification by flash column chromatography using silica gel, eluting with 30% diethyl ether in petroleum ether (40–60) gave (2R,5E)-2-(tert-butoxycarbonylamino)-8-cyclohexyl-4-oxodec-5-ene (10h) as a colourless oil (0.037 g, 79%). νmax/cm−1 (neat) 3341 (NH), 2924 (CH), 1694 (CO), 1512, 1366, 1246, 1169, 1053; [α]24D +5.1 (c 0.9, CHCl3); δH (400 MHz, CDCl3) 0.80–0.96 (2H, m, CH2), 1.09–1.28 (8H, m, 1-H3, 8-CH, 2 × CH2), 1.34 (2H, q, J 6.9 Hz, 8-H2), 1.43 (9H, s, 3 × CH3), 1.60–1.78 (4H, m, 2 × CH2), 2.22 (2H, qd, J 6.9, 1.5 Hz, 7-H2), 2.63 (1H, dd, J 15.8, 6.6 Hz, 3-HH), 2.85 (1H, dd, J 15.8, 4.8 Hz, 3-HH), 3.97–4.07 (1H, m, 2-H), 4.94 (1H, br s, NH), 6.08 (1H, dd, J 15.9, 1.5 Hz, 5-H), 6.85 (1H, dd, J 15.9, 6.9 Hz, 6-H); δC (101 MHz, CDCl3) 20.7 (CH3), 26.4 (2 × CH2), 26.7 (CH2), 28.6 (3 × CH3), 30.1 (CH2), 33.3 (2 × CH2), 35.8 (CH2), 37.3 (CH), 43.9 (CH), 45.8 (CH2), 79.3 (C), 130.6 (CH), 149.0 (CH), 155.3 (C), 199.3 (C); m/z (ESI) 346.2343 (MNa+. C19H33NNaO3 requires 346.2353).
(2R,5E)-2-(tert-Butoxycarbonylamino)-4-oxo-8-phenyloct-5-ene (10i)
The reaction was carried out according to the above procedure for the synthesis of (2R,5E)-2-(tert-butoxycarbonylamino)-4-oxonona-5-ene (10a) using (4R)-4-(tert-butoxycarbonylamino)-1-(dimethyloxyphosphoryl)pentan-2-one (9) (0.349 g, 1.13 mmol) and hydrocinnamaldehyde (0.300 mL, 2.26 mmol) for 48 h. Purification by flash column chromatography using silica gel, eluting with 20% ethyl acetate in petroleum ether (40–60) gave (2R,5E)-2-(tert-butoxycarbonylamino)-4-oxo-8-phenyloct-5-ene (10i) (0.288 g, 80%) as a pale yellow oil. νmax/cm−1 (neat) 3353 (NH), 2976 (CH), 1692 (CO), 1496 (CC), 1247, 1221, 1054; [α]23D +4.1 (c 1.0, CHCl3); δH (400 MHz, CDCl3) 1.17 (3H, d, J 6.7 Hz, 1-H3), 1.43 (9H, s, 3 × CH3), 2.53 (2H, q, J 6.8 Hz, 7-H2), 2.60 (1H, dd, J 15.7, 7.8 Hz, 3-HH), 2.77 (2H, t, J 6.8 Hz, 8-H2), 2.84 (1H, dd, J 15.7, 4.5 Hz, 3-HH), 3.95–4.08 (1H, m, 2-H), 4.99 (1H, br s, NH), 6.09 (1H, d, J 15.9 Hz, 5-H), 6.87 (1H, dt, J 15.9, 6.8 Hz, 6-H), 7.14–7.23 (3H, m, 3 × ArH), 7.25–7.32 (2H, m, 2 × ArH); δC (101 MHz, CDCl3) 20.5 (CH3), 28.5 (3 × CH3), 34.2 (CH2), 34.4 (CH2), 43.7 (CH), 45.9 (CH2), 79.2 (C), 126.3 (CH), 128.4 (2 × CH), 128.6 (2 × CH), 131.1 (CH), 140.7 (C), 147.0 (CH), 155.2 (C), 199.0 (C); m/z (ESI) 340.1868 (MNa+. C19H27NNaO3 requires 340.1883).
(2R,5E)-2-(tert-Butoxycarbonylamino)-8-methyl-4-oxonon-5-ene (10j)
The reaction was carried out according to the above procedure for the synthesis of (2R,5E)-2-(tert-butoxycarbonylamino)-4-oxonona-5-ene (10a) using (4R)-4-(tert-butoxycarbonylamino)-1-(dimethyloxyphosphoryl)pentan-2-one (9) (0.141 g, 0.455 mmol) and isovaleraldehyde (0.097 mL, 0.910 mmol). Purification by flash column chromatography using silica gel, eluting with 40% diethyl ether in petroleum ether (40–60) gave (2R,5E)-2-(tert-butoxycarbonylamino)-8-methyl-4-oxonon-5-ene (10j) (0.080 g, 70%) as a colourless oil. νmax/cm−1 (neat) 3356 (NH), 2963 (CH), 1690 (CO), 1504, 1366, 1165, 1250, 1049; [α]29D +7.5 (c 0.5, CHCl3); δH (400 MHz, CDCl3) 0.91 (6H, d, J 6.7 Hz, 8-CH3 and 9-H3), 1.18 (3H, d, J 6.8 Hz, 1-H3), 1.41 (9H, s, 3 × CH3), 1.67–1.83 (1H, m, 8-H), 2.09 (2H, td, J 7.5, 1.3 Hz, 7-H2), 2.62 (1H, dd, J 15.9, 6.6 Hz, 3-HH), 2.84 (1H, dd, J 15.9, 4.8 Hz, 3-HH), 3.94–4.08 (1H, m, 2-H), 4.95 (1H, br s, NH), 6.06 (1H, dt, J 15.7, 1.3 Hz, 5-H), 6.81 (1H, dt, J 15.7, 7.5, 6-H); δC (101 MHz, CDCl3) 20.6 (CH3), 22.5 (2 × CH3), 28.0 (CH), 28.5 (3 × CH3), 41.9 (CH2), 43.9 (CH), 45.9 (CH2), 79.3 (C), 131.8 (CH), 147.3 (CH), 155.3 (C), 199.1 (C); m/z (ESI) 292.1873 (MNa+. C15H27NNaO3 requires 292.1883).
(2R,5E)-2-(tert-Butoxycarbonylamino)-7-methyl-4-oxooct-5-ene (10k)
The reaction was carried out according to the above procedure for the synthesis of (2R,5E)-2-(tert-butoxycarbonylamino)-4-oxonona-5-ene (10a) using (4R)-4-(tert-butoxycarbonylamino)-1-(dimethyloxyphosphoryl)pentan-2-one (9) (0.160 g, 0.517 mmol) and isobutyraldehyde (0.095 mL, 1.04 mmol). Purification by flash column chromatography using silica gel, eluting with 30% diethyl ether in petroleum ether (40–60) gave (2R,5E)-2-(tert-butoxycarbonylamino)-7-methyl-4-oxooct-5-ene (10k) (0.112 g, 85%) as a colourless oil. νmax/cm−1 (neat) 3356 (NH), 2970 (CH), 1690 (CO), 1512, 1366, 1250, 1165, 1049; [α]29D +9.4 (c 1.2, CHCl3); δH (400 MHz, CDCl3) 1.02 (6H, d, J 6.8 Hz, 7-CH3 and 8-H3), 1.15 (3H, d, J 6.8 Hz, 1-H3), 1.38 (9H, s, 3 × CH3), 2.35–2.47 (1H, m, 7-H), 2.60 (1H, dd, J 16.0, 6.6 Hz, 3-HH), 2.81 (1H, dd, J 16.0, 4.8 Hz, 3-HH), 3.90–4.05 (1H, m, 2-H), 4.97 (1H, br s, NH), 5.99 (1H, dd, J 16.0, 1.2 Hz, 5-H), 6.76 (1H, dd, J 16.0, 6.7 Hz, 6-H); δC (101 MHz, CDCl3) 20.6 (CH3), 21.3 (2 × CH3), 28.5 (3 × CH3), 31.2 (CH), 43.7 (CH), 45.8 (CH2), 79.1 (C), 127.8 (CH), 154.3 (CH), 155.2 (C), 199.4 (C); m/z (ESI) 278.1722 (MNa+. C14H25NNaO3 requires 278.1727).
(2R,5E)-2-(tert-Butoxycarbonylamino)-4-oxo-6-phenylhex-5-ene (10l)
The reaction was carried out according to the above procedure for the synthesis of (2R,5E)-2-(tert-butoxycarbonylamino)-4-oxonona-5-ene (10a) using (4R)-4-(tert-butoxycarbonylamino)-1-(dimethyloxyphosphoryl)pentan-2-one (9) (0.251 g, 0.810 mmol) and benzaldehyde (0.160 mL, 1.62 mmol) for 48 h. Purification by flash column chromatography using silica gel, eluting with 20% ethyl acetate in petroleum ether (40–60) gave (2R,5E)-2-(tert-butoxycarbonylamino)-4-oxo-6-phenylhex-5-ene (10l) (0.227 g, 97%) as a white solid. Mp 59–62 °C; νmax/cm−1 (neat) 3345 (NH), 2976 (CH), 1687 (CO), 1655 (CO), 1608 (CC), 1495, 1365, 1247, 1166; [α]23D +10.0 (c 1.0, CHCl3); δH (400 MHz, CDCl3) 1.25 (3H, d, J 6.8 Hz, 1-H3), 1.43 (9H, s, 3 × CH3), 2.77 (1H, dd, J 15.8, 6.8 Hz, 3-HH), 3.00 (1H, dd, J 15.8, 4.6 Hz, 3-HH), 4.05–4.18 (1H, m, 2-H), 5.04 (1H, br s, NH), 6.73 (1H, d, J 16.2 Hz, 5-H), 7.36–7.41 (3H, m, 3 × ArH), 7.52–7.54 (2H, m, 2 × ArH), 7.56 (1H, d, J 16.2 Hz, 6-H); δC (101 MHz, CDCl3) 20.6 (CH3), 28.4 (3 × CH3), 43.8 (CH), 46.6 (CH2), 79.2 (C), 126.4 (CH), 128.4 (2 × CH), 129.0 (2 × CH), 130.6 (CH), 134.4 (C), 143.2 (CH), 155.2 (C), 198.9 (C); m/z (ESI) 312.1558 (MNa+. C17H23NNaO3 requires 312.1570).
tert-Butyl (2R,6R)-2-methyl-4-oxo-6-propylpiperidine-1-carboxylate (11a)
(2R,5E)-2-(tert-Butoxycarbonylamino)-4-oxonona-5-ene (10a) (0.0756 g, 0.296 mmol) was dissolved in methanol (7.4 mL) and cooled to −78 °C. 2 M hydrochloric acid in diethyl ether (0.148 mL, 0.296 mmol) was added and the solution stirred at this temperature for 1 h and, then at room temperature for 1 h. Upon completion, the mixture was concentrated in vacuo. Purification by flash column chromatography using silica gel, eluting with 30% diethyl ether in petroleum ether (40–60) gave tert-butyl (2R,6R)-2-methyl-4-oxo-6-propylpiperidine-1-carboxylate (11a) (0.0369 g, 49%) as a colourless oil. νmax/cm−1 (neat) 2964 (CH), 1726 (CO), 1688 (CO), 1389, 1174, 1101; [α]22D +151.7 (c 1.0, CHCl3); δH (400 MHz, CDCl3) 0.90 (3H, t, J 7.2 Hz, 3′′-H3), 1.20–1.39 (6H, m, 1′-H3, 1′′-HH and 2′′-H2), 1.49 (9H, s, 3 × CH3), 1.65–1.78 (1H, m, 1′′-HH), 2.35 (1H, dd, J 17.7, 2.0 Hz, 3-HH), 2.52 (1H, dd, J 17.9, 2.0 Hz, 5-HH), 2.70 (1H, ddd, J 17.9, 6.3, 1.4 Hz, 5-HH), 2.81 (1H, dd, J 17.7, 6.3 Hz, 3-HH), 4.10–4.22 (1H, m, 6-H), 4.29–4.40 (1H, m, 2-H); δC (101 MHz, CDCl3) 14.0 (CH3), 20.1 (CH2), 22.9 (CH3), 28.6 (3 × CH3), 39.4 (CH2), 41.4 (CH2), 44.7 (CH2), 46.6 (CH), 51.0 (CH), 80.0 (C), 154.7 (C), 208.3 (C); m/z (ESI) 278.1730 (MNa+. C14H25NNaO3 requires 278.1727).
tert-Butyl (2R,6R)-6-butyl-2-methyl-4-oxopiperidine-1-carboxylate (11b)
The reaction was carried out according to the above procedure for the synthesis of tert-butyl (2R,6R)-2-methyl-4-oxo-6-propylpiperidine-1-carboxylate (11a) using (2R,5E)-2-(tert-butoxycarbonylamino)-4-oxodec-5-ene (10b) (0.0616 g, 0.229 mmol). Purification by flash column chromatography using silica gel, eluting with 20% diethyl ether in petroleum ether (40–60) gave tert-butyl (2R,6R)-6-butyl-2-methyl-4-oxopiperidine-1-carboxylate (11b) (0.0320 g, 52%) as a colourless oil. νmax/cm−1 (neat) 2961 (CH), 1727 (CO), 1691 (CO), 1390, 1175; [α]20D +131.8 (c 0.2, CHCl3); δH (400 MHz, CDCl3) 0.88 (3H, t, J 7.0 Hz, 4′′-H3), 1.17–1.39 (8H, m, 1′-H3, 1′′-HH, 2′′-H2 and 3′′-H2), 1.49 (9H, s, 3 × CH3), 1.68–1.86 (1H, m, 1′′-HH), 2.35 (1H, dd, J 17.7, 1.9 Hz, 3-HH), 2.52 (1H, dd, J 17.9, 2.1 Hz, 5-HH), 2.72 (1H, ddd, J 17.9, 6.2, 1.4 Hz, 5-HH), 2.81 (1H, dd, J 17.7, 6.4 Hz, 3-HH), 4.04–4.24 (1H, m, 6-H), 4.30–4.44 (1H, m, 2-H); δC (101 MHz, CDCl3) 14.0 (CH3), 22.5 (CH2), 22.7 (CH3), 28.5 (3 × CH3), 28.9 (CH2), 36.9 (CH2), 41.3 (CH2), 44.6 (CH2), 46.5 (CH), 51.2 (CH), 79.9 (C), 154.6 (C), 208.0 (C); m/z (ESI) 292.1886 (MNa+. C15H27NNaO3 requires 292.1889).
tert-Butyl (2R,6R)-2-methyl-4-oxo-6-pentylpiperidine-1-carboxylate (11c)
The reaction was carried out according to the above procedure for the synthesis of tert-butyl (2R,6R)-2-methyl-4-oxo-6-propylpiperidine-1-carboxylate (11a) using (2R,5E)-2-(tert-butoxycarbonylamino)-4-oxoundec-5-ene (10c) (0.0852 g, 0.301 mmol). Purification by flash column chromatography using silica gel, eluting with 20% diethyl ether in petroleum ether (40–60) gave tert-butyl (2R,6R)-2-methyl-4-oxo-6-pentylpiperidine-1-carboxylate (11c) (0.0402 g, 47%) as a colourless oil. νmax/cm−1 (neat) 2932 (CH), 1728 (CO), 1690 (CO), 1366, 1173; [α]25D +139.9 (c 0.4, CHCl3); δH (500 MHz, CDCl3) 0.86 (3H, t, J 6.8 Hz, 5′′-H3), 1.17–1.35 (10H, m, 1′-H3, 1′′-HH, 2′′-H2, 3′′-H2 and 4′′-H2), 1.49 (9H, s, 3 × CH3), 1.67–1.80 (1H, m, 1′′-HH), 2.36 (1H, dd, J 17.7, 1.9 Hz, 3-HH), 2.53 (1H, dd, J 17.9, 2.0 Hz, 5-HH), 2.71 (1H, ddd, J 17.9, 6.2, 1.3 Hz, 5-HH), 2.80 (1H, dd, J 17.7, 6.4 Hz, 3-HH), 4.05–4.22 (1H, m, 6-H), 4.25–4.40 (1H, m, 2-H); δC (126 MHz, CDCl3) 14.1 (CH3), 22.7 (CH2), 22.9 (CH3), 26.6 (CH2), 28.6 (3 × CH3), 31.7 (CH2), 37.3 (CH2), 41.4 (CH2), 44.7 (CH2), 46.6 (CH), 51.3 (CH), 80.0 (C), 154.7 (C), 208.3 (C); m/z (ESI) 306.2034 (MNa+. C16H29NNaO3 requires 306.2040).
tert-Butyl (2R,6R)-2-methyl-6-octyl-4-oxopiperidine-1-carboxylate (11d)
The reaction was carried out according to the above procedure for the synthesis of tert-butyl (2R,6R)-2-methyl-4-oxo-6-propylpiperidine-1-carboxylate (11a) using (2R,5E)-2-(tert-butoxycarbonylamino)-4-oxotetraadec-5-ene (10d) (0.0977 g, 0.300 mmol). Purification by flash column chromatography using silica gel, eluting with 20% diethyl ether in petroleum ether (40–60) gave tert-butyl (2R,6R)-2-methyl-6-octyl-4-oxopiperidine-1-carboxylate (11d) (0.0495 g, 51%) as a colourless oil. νmax/cm−1 (neat) 2924 (CH), 1728 (CO), 1690 (CO), 1366, 1172; [α]30D +158.2 (c 0.6, CHCl3); δH (400 MHz, CDCl3) 0.87 (3H, t, J 6.9 Hz, 8′′-H3), 1.15–1.35 (16H, m, 1′-H3, 1′′-HH, 2′′-H2, 3′′-H2, 4′′-H2, 5′′-H2, 6′′-H2, and 7′′-H2), 1.49 (9H, s, 3 × CH3), 1.65–1.80 (1H, m, 1′′-HH), 2.36 (1H, dd, J 17.8, 1.9 Hz, 3-HH), 2.53 (1H, dd, J 17.9, 2.0 Hz, 5-HH), 2.72 (1H, ddd, J 17.9, 6.2, 1.3 Hz, 5-HH), 2.81 (1H, dd, J 17.8, 6.4 Hz, 3-HH), 4.05–4.20 (1H, m, 6-H), 4.28–4.45 (1H, m, 2-H); δC (101 MHz, CDCl3) 14.2 (CH3), 22.8 (CH2), 22.9 (CH3), 26.9 (CH2), 28.6 (3 × CH3), 29.3 (CH2), 29.6 (CH2), 29.7 (CH2), 32.0 (CH2), 37.3 (CH2), 41.4 (CH2), 44.7 (CH2), 46.6 (CH), 51.3 (CH), 80.0 (C), 154.7 (C), 208.3 (C); m/z (ESI) 348.2515 (MNa+. C19H35NNaO3 requires 348.2509).
tert-Butyl (2R,6R)-2-methyl-6-nonyl-4-oxopiperidine-1-carboxylate (11e)
The reaction was carried out according to the above procedure for the synthesis of tert-butyl (2R,6R)-2-methyl-4-oxo-6-propylpiperidine-1-carboxylate (11a) using (2R,5E)-2-(tert-butoxycarbonylamino)-4-oxopentadec-5-ene (10e) (0.0996 g, 0.148 mmol). Purification by flash column chromatography using silica gel, eluting with 20% diethyl ether in petroleum ether (40–60) gave tert-butyl (2R,6R)-2-methyl-6-nonyl-4-oxopiperidine-1-carboxylate (11e) (0.0444 g, 55%) as a colourless oil. νmax/cm−1 (neat) 2925 (CH), 1726 (CO), 1691 (CO), 1389, 1175, 1094; [α]23D +102.4 (c 1.1, CHCl3); δH (400 MHz, CDCl3) 0.87 (3H, t, J 6.8 Hz, 9′′-H3), 1.16–1.36 (18H, m, 1′-H3, 1′′-HH, 2′′-H2, 3′′-H2, 4′′-H2, 5′′-H2, 6′′-H2, 7′′-H2 and 8′′-H2), 1.49 (9H, s, 3 × CH3), 1.68–1.80 (1H, m, 1′′-HH), 2.35 (1H, dd, J 17.7, 1.8 Hz, 3-HH), 2.52 (1H, dd, J 18.0, 1.8 Hz, 5-HH), 2.71 (1H, ddd, J 18.0, 5.5, 0.8 Hz, 5-HH), 2.80 (1H, dd, J 17.7, 6.4 Hz, 3-HH), 4.08–4.19 (1H, m, 6-H), 4.29–4.40 (1H, m, 2-H); δC (101 MHz, CDCl3) 14.1 (CH3), 22.7 (CH2), 22.7 (CH3), 26.8 (CH2), 28.5 (3 × CH3), 29.3 (CH2), 29.4 (CH2), 29.5 (CH2), 29.6 (CH2), 31.9 (CH2), 37.2 (CH2), 41.3 (CH2), 44.6 (CH2), 46.5 (CH), 51.2 (CH), 79.9 (C), 154.5 (C), 208.2 (C); m/z (ESI) 362.2662 (MNa+. C20H37NNaO3 requires 362.2666).
tert-Butyl (2R,6R)-2-methyl-4-oxo-6-undecylpiperidine-1-carboxylate (11f)
The reaction was carried out according to the above procedure for the synthesis of tert-butyl (2R,6R)-2-methyl-4-oxo-6-propylpiperidine-1-carboxylate (11a) using (2R,5E)-2-(tert-butoxycarbonylamino)-4-oxoheptadec-5-ene (10f) (0.113 g, 0.307 mmol). Purification by flash column chromatography using silica gel, eluting with 20% diethyl ether in petroleum ether (40–60) gave tert-butyl (2R,6R)-2-methyl-4-oxo-6-undecylpiperidine-1-carboxylate (11f) (0.0551 g, 49%) as a colourless oil. νmax/cm−1 (neat) 2924 (CH), 1726 (CO), 1691 (CO), 1389, 1174, 1095; [α]24D +92.7 (c 1.0, CHCl3); δH (400 MHz, CDCl3) 0.88 (3H, t, J 6.9 Hz, 11′′-H3), 1.18–1.38 (22H, m, 1′-H3, 1′′-HH, 2′′-H2, 3′′-H2, 4′′-H2, 5′′-H2, 6′′-H2, 7′′-H2, 8′′-H2, 9′′-H2 and 10′′-H2), 1.49 (9H, s, 3 × CH3), 1.66–1.80 (1H, m, 1′′-HH), 2.36 (1H, dd, J 17.7, 1.9 Hz, 3-HH), 2.53 (1H, dd, J 17.9, 2.0 Hz, 5-HH), 2.71 (1H, ddd, J 17.9, 6.2, 1.3 Hz, 5-HH), 2.81 (1H, dd, J 17.7, 6.5 Hz, 3-HH), 4.07–4.23 (1H, m, 6-H), 4.27–4.43 (1H, m, 2-H); δC (101 MHz, CDCl3) 14.3 (CH3), 22.8 (CH2), 22.9 (CH3), 26.9 (CH2), 28.7 (3 × CH3), 29.5 (CH2), 29.6 (CH2), 29.7 (3 × CH2), 29.8 (CH2) 32.1 (CH2), 37.3 (CH2), 41.4 (CH2), 44.7 (CH2), 46.6 (CH), 51.3 (CH), 80.0 (C), 154.7 (C), 208.3 (C); m/z (ESI) 390.2963 (MNa+. C22H41NNaO3 requires 390.2979).
tert-Butyl (2R,6R)-6-(4′′-chlorobutyl)-2-methyl-4-oxopiperidine-1-carboxylate (11g)
The reaction was carried out according to the above procedure for the synthesis of tert-butyl (2R,6R)-2-methyl-4-oxo-6-propylpiperidine-1-carboxylate (11a) using (2R,5E)-2-(tert-butoxycarbonylamino)-10-chloro-4-oxodec-5-ene (10g) (0.0484 g, 0.0159 mmol). Purification by flash column chromatography using silica gel, eluting with 20% diethyl ether in petroleum ether (40–60) gave tert-butyl (2R,6R)-6-(4′′-chlorobutyl)-2-methyl-4-oxopiperidine-1-carboxylate (11g) (0.0269 g, 56%) as a white solid. Mp 58–59 °C; νmax/cm−1 (neat) 2975 (CH), 1725 (CO), 1688 (CO), 1390, 1366, 1174, 1097; [α]22D +120.1 (c 1.0, CHCl3); δH (400 MHz, CDCl3) 1.25 (3H, d, J 6.6 Hz, 1′-H3), 1.30–1.47 (3H, m, 1-HH and 2′′-H2), 1.49 (9H, s, 3 × CH3), 1.69–1.84 (3H, m, 3′′-H2 and 1′′-HH), 2.37 (1H, dd, J 17.8, 1.9 Hz, 3-HH), 2.52 (1H, dd, J 18.0, 2.0 Hz, 5-HH), 2.73 (1H, ddd, J 18.0, 6.0, 0.8 Hz, 5-HH), 2.78 (1H, dd, J 17.8, 6.4 Hz, 3-HH), 3.51 (2H, t, J 6.5 Hz, 4′′-H2), 4.09–4.24 (1H, m, 6-H), 4.26–4.43 (1H, m, 2-H); δC (101 MHz, CDCl3) 22.8 (CH3), 24.1 (CH2), 28.6 (3 × CH3), 32.3 (CH2), 36.5 (CH2), 41.5 (CH2), 44.7 (CH2), 44.8 (CH2), 46.7 (CH), 51.0 (CH), 80.2 (C), 154.7 (C), 207.9 (C); m/z (ESI) 326.1480 (MNa+. C15H2635ClNNaO3 requires 326.1493).
tert-Butyl (2R,6R)-6-(2′′-cyclohexylethyl)-2-methyl-4-oxopiperidine-1-carboxylate (11h)
The reaction was carried out according to the above procedure for the synthesis of tert-butyl (2R,6R)-2-methyl-4-oxo-6-propylpiperidine-1-carboxylate (11a) using (2R,5E)-2-(tert-butoxycarbonylamino)-8-cyclohexyl-4-oxodec-5-ene (10h) (0.0269 g, 0.0830 mmol). Purification by flash column chromatography using silica gel, eluting with 20% diethyl ether in petroleum ether (40–60) gave tert-butyl (2R,6R)-6-(2′′-cyclohexylethyl)-2-methyl-4-oxopiperidine-1-carboxylate (11h) (0.0149 g, 55%) as a colourless oil. νmax/cm−1 (neat) 2924 (CH), 1721 (CO), 1690 (CO), 1389, 1366, 1173, 1096; [α]25D +105.9 (c 0.4, CHCl3); δH (400 MHz, CDCl3) 0.75–0.95 (2H, m, CH2), 1.05–1.38 (11H, m, 1′-H3, 1′′-HH, 2′′-H2, 3′′-H, and 2 × CH2), 1.49 (9H, s, 3 × CH3), 1.60–1.84 (5H, m, 1′′-HH and 2 × CH2), 2.36 (1H, dd, J 17.8, 1.9 Hz, 3-HH), 2.52 (1H, dd, J 17.9, 2.0 Hz, 5-HH), 2.71 (1H, ddd, J 17.9, 6.2, 1.2 Hz, 5-HH), 2.80 (1H, dd, J 17.8, 6.4 Hz, 3-HH), 4.03–4.17 (1H, m, 6-H), 4.28–4.44 (1H, m, 2-H); δC (101 MHz, CDCl3) 22.8 (CH3), 26.4 (2 × CH2), 26.7 (CH2), 28.7 (3 × CH3), 33.4 (CH2), 33.6 (CH2), 34.6 (CH2), 34.7 (CH2), 37.7 (CH), 41.4 (CH2), 44.7 (CH2), 46.6 (CH), 51.7 (CH), 80.0 (C), 154.7 (C), 208.3 (C); m/z (ESI) 346.2345 (MNa+. C19H33NNaO3 requires 346.2353).
tert-Butyl (2R,6R)-2-methyl-4-oxo-6-(2′′-phenylethyl)piperidine-1-carboxylate (11i)
The reaction was carried out according to the above procedure for the synthesis of tert-butyl (2R,6R)-2-methyl-4-oxo-6-propylpiperidine-1-carboxylate (11a) using (2R,5E)-2-(tert-butoxycarbonylamino)-4-oxo-8-phenyloct-5-ene (10i) (0.090 g, 0.284 mmol). Purification by flash column chromatography using silica gel, eluting with 20% ethyl acetate in petroleum ether (40–60) gave tert-butyl (2R,6R)-2-methyl-4-oxo-6-(2′′-phenylethyl)piperidine-1-carboxylate (11i) (0.045 g, 51%) as a white solid. Mp 76–78 °C; νmax/cm−1 (neat) 2973 (CH), 1724 (CO), 1687 (CO), 1389, 1366, 1173, 1096; [α]18D +97.7 (c 1.0, CHCl3); δH (400 MHz, CDCl3) 1.27 (3H, d, J 6.7 Hz, 1′-H3), 1.49 (9H, s, 3 × CH3), 1.58–1.70 (1H, m, 1′′-HH), 2.04–2.16 (1H, m, 1′′-HH), 2.38 (1H, dd, J 17.8, 2.0 Hz, 3-HH), 2.54–2.69 (3H, m, 5-HH and 2′′-H2), 2.77 (1H, dd, J 17.5, 5.6 Hz, 5-HH), 2.82 (1H, dd, J 17.8, 6.8 Hz, 3-HH), 4.20–4.30 (1H, m, 6-H), 4.31–4.43 (1H, m, 2-H), 7.12–7.32 (5H, m, Ph); δC (101 MHz, CDCl3) 22.7 (CH3), 28.5 (3 × CH3), 33.2 (CH2), 39.0 (CH2), 41.4 (CH2), 44.6 (CH2), 46.6 (CH), 50.9 (CH), 80.1 (C), 126.1 (CH), 128.3 (2 × CH), 128.5 (2 × CH), 141.0 (C), 154.5 (C), 207.7 (C); m/z (ESI) 340.1875 (MNa+. C19H27NNaO3 requires 340.1883).
tert-Butyl (2R,6R)-2-methyl-6-(2′′-methylpropyl)-4-oxopiperidine-1-carboxylate (11j) and tert-butyl (2R,6S)-2-methyl-6-(2′′-methylpropyl)-4-oxopiperidine-1-carboxylate (12j)
The reaction was carried out according to the above procedure for the synthesis of tert-butyl (2R,6R)-2-methyl-4-oxo-6-propylpiperidine-1-carboxylate (11a) using (2R,5E)-2-(tert-butoxycarbonylamino)-8-methyl-4-oxonon-5-ene (10j) (0.0804 g, 0.298 mmol). Purification by flash column chromatography using silica gel, eluting with 20% diethyl ether in petroleum ether (40–60) gave tert-butyl (2R,6R)-2-methyl-6-(2′′-methylpropyl)-4-oxopiperidine-1-carboxylate (11j) (0.032 g, 40%) and tert-butyl (2R,6S)-2-methyl-6-(2′′-methylpropyl)-4-oxopiperidine-1-carboxylate (12j) (0.032 g, 40%) as colourless oils. Data for tert-butyl (2R,6R)-2-methyl-6-(2′′-methylpropyl)-4-oxopiperidine-1-carboxylate (11j): νmax/cm−1 (neat) 2960 (CH), 1726 (CO), 1688 (CO), 1389, 1369, 1175; [α]28D +34.4 (c 0.2, CHCl3); δH (500 MHz, CDCl3) 0.89 (3H, d, J 6.3 Hz, 3′′-H3), 0.92 (3H, d, J 6.3 Hz, 2′′-CH3), 1.26 (3H, d, J 6.7 Hz, 1′-H3), 1.27–1.33 (1H, m, 2′′-H), 1.50 (9H, s, 3 × CH3), 1.52–1.58 (2H, m, 1′′-H2), 2.36 (1H, dd, J 17.8, 2.0 Hz, 3-HH), 2.52 (1H, dd, J 17.9, 2.0 Hz, 5-HH), 2.72 (1H, ddd, J 17.9, 6.1, 1.0 Hz, 5-HH), 2.82 (1H, dd, J 17.8, 6.5 Hz, 3-HH), 4.22–4.30 (1H, m, 6-H), 4.31–4.39 (1H, m, 2-H); δC (126 MHz, CDCl3) 21.4 (CH3), 22.9 (CH), 23.9 (CH3), 25.5 (CH3), 28.7 (3 × CH3), 41.5 (CH2), 44.7 (CH2), 45.8 (CH2), 46.7 (CH), 49.4 (CH), 80.1 (C), 154.6 (C), 208.2 (C); m/z (ESI) 292.1877 (MNa+. C15H27NNaO3 requires 292.1883). Data for tert-butyl (2R,6S)-2-methyl-6-(2′′-methylpropyl)-4-oxopiperidine-1-carboxylate (12j): νmax/cm−1 (neat) 2961 (CH), 1721 (CO), 1686 (CO), 1352, 1171, 1125; [α]28D −4.0 (c 0.3, CHCl3); δH (500 MHz, CDCl3) 0.91 (3H, d, J 6.5 Hz, 3′′-H3), 0.94 (3H, d, J 6.5 Hz, 2′′-CH3), 1.26 (3H, d, J 7.0 Hz, 1′-H3), 1.33–1.44 (2H, m, 1′′-H2), 1.48 (9H, s, 3 × CH3), 1.51–1.59 (1H, m, 2′′-H), 2.25–2.33 (2H, m, 3-HH and 5-HH), 2.59–2.73 (2H, m, 3-HH and 5-HH), 4.58–4.82 (2H, m, 2-H and 6-H); δC (126 MHz, CDCl3) 22.2 (CH3), 23.0 (CH), 23.2 (CH3), 25.4 (CH3), 28.6 (3 × CH3), 44.1 (CH2), 45.6 (CH2), 46.0 (CH2), 48.4 (CH), 50.8 (CH), 80.4 (C), 154.8 (C), 208.8 (C); m/z (ESI) 292.1877 (MNa+. C15H27NNaO3 requires 292.1883).
tert-Butyl (2R,6R)-2-methyl-6-(1′′-methylethyl)-4-oxopiperidine-1-carboxylate (11k) and tert-butyl (2R,6S)-2-methyl-6-(1′′-methylethyl)-4-oxopiperidine-1-carboxylate (12k)
The reaction was carried out according to the above procedure for the synthesis of tert-butyl (2R,6R)-2-methyl-4-oxo-6-propylpiperidine-1-carboxylate (11a) using (2R,5E)-2-(tert-butoxycarbonylamino)-7-methyl-4-oxooct-5-ene (10k) (0.103 g, 0.403 mmol). Purification by flash column chromatography using silica gel, eluting with 20% diethyl ether in petroleum ether (40–60) gave tert-butyl (2R,6R)-2-methyl-6-(1′′-methylethyl)-4-oxopiperidine-1-carboxylate (11k) (0.027 g, 27%) and tert-butyl (2R,6S)-2-methyl-6-(1′′-methylethyl)-4-oxopiperidine-1-carboxylate (12k) (0.027 g, 27%) as colourless oils. Data for tert-butyl (2R,6R)-2-methyl-6-(1′′-methylethyl)-4-oxopiperidine-1-carboxylate (11k): νmax/cm−1 (neat) 2968 (CH), 1724 (CO), 1688 (CO), 1381, 1366, 1354, 1175; [α]27D +171.1 (c 0.2, CHCl3); δH (500 MHz, CDCl3) 0.92 (3H, d, J 6.5 Hz, 2′′-H3), 0.97 (3H, d, J 6.5 Hz, 1′′-CH3), 1.26 (3H, d, J 6.5 Hz, 1′-H3), 1.49 (9H, s, 3 × CH3), 1.60–1.72 (1H, m, 1′′-H), 2.33 (2H, dd, J 17.5, 2.2 Hz, 3-HH), 2.59 (1H, dd, J 18.0, 2.6 Hz, 5-HH), 2.66 (1H, dd, J 18.0, 5.8 Hz, 5-HH), 2.79 (1H, dd, J 17.5, 6.5 Hz, 3-HH), 3.95–4.10 (1H, m, 6-H), 4.31 (1H, quin.d, J 6.5, 2.2 Hz, 2-H); δC (126 MHz, CDCl3) 19.3 (CH3), 20.4 (CH3), 22.8 (CH3), 28.6 (3 × CH3), 35.1 (CH), 41.5 (CH2), 44.8 (CH2), 47.0 (CH), 56.3 (CH), 80.1 (C), 155.5 (C), 208.6 (C); m/z (ESI) 278.1722 (MNa+. C14H25NNaO3 requires 278.1727). Data for tert-butyl (2R,6S)-2-methyl-6-(1′′-methylethyl)-4-oxopiperidine-1-carboxylate (12k): νmax/cm−1 (neat) 2972 (CH), 1718 (CO), 1687 (CO), 1365, 1172, 1120, 1066; [α]23D −47.6 (c 0.5, CHCl3); δH (500 MHz, CDCl3) 0.93 (3H, d, J 6.5 Hz, 2′′-H3), 0.97 (3H, d, J 6.5 Hz, 1′′-CH3), 1.30 (3H, d, J 6.8 Hz, 1′-H3), 1.48 (9H, s, 3 × CH3), 1.76–1.84 (1H, m, 1′′-H), 2.29 (1H, ddd, J 15.5, 5.4, 0.9 Hz, 3-HH), 2.53–2.57 (2H, m, 5-H2), 2.67 (1H, dd, J 15.5, 7.7 Hz, 3-HH), 4.15–4.35 (1H, m, 6-H), 4.52–4.75 (1H, m, 2-H); δC (126 MHz, CDCl3) 20.2 (CH3), 20.4 (CH3), 22.6 (CH3), 28.4 (3 × CH3), 33.0 (CH), 42.0 (CH2), 45.5 (CH2), 48.1 (CH), 58.5 (CH), 80.2 (C), 155.3 (C), 208.8 (C); m/z (ESI) 278.1720 (MNa+. C14H25NNaO3 requires 278.1727).
(+)-Myrtine (1)10b
To a solution of tert-butyl (2R,6R)-6-(4′′-chlorobutyl)-2-methyl-4-oxopiperidine-1-carboxylate (11g) (0.049 g, 0.16 mmol) in tetrahydrofuran (2 mL) was added aqueous 2 M hydrochloric acid solution. The reaction mixture was stirred at room temperature for 4 h. The reaction mixture was concentrated in vacuo to afford a yellow residue. This was dissolved in acetone (5 mL) and saturated sodium hydrogen carbonate solution (3 mL) and water (2 mL) were added. The reaction mixture was stirred at room temperature for 48 h. The reaction mixture was poured onto saturated sodium hydrogen carbonate solution (50 mL), which was extracted with dichloromethane (3 × 50 mL). The combined organic layers were washed with brine (50 mL), dried (MgSO4) and concentrated in vacuo to give the crude product as a yellow oil. Purification by flash column chromatography using silica gel, eluting with 4% methanol in dichloromethane gave (+)-myrtine (1) as a pale yellow oil (0.017 g, 62%). [α]24D +8.9 (c 0.3, CHCl3), lit.10b [α]22D +10.5 (c 0.9, CHCl3); δH (500 MHz, CDCl3) 0.95 (3H, d, J 6.8 Hz, 1′-H3), 1.15–1.35 (2H, m, 2′′-HH and 3′′-HH), 1.52–1.76 (4H, m, 1′′-H2, 2′′-HH and 3′′-HH), 2.14–2.28 (3H, m, 3-HH and 4′′-H2), 2.46 (1H, td, J 11.6, 2.8 Hz, 5-HH), 2.60–2.69 (1H, m, 6-H), 2.74–2.80 (1H, m, 5-HH), 2.83 (1H, dd, J 13.4, 6.8 Hz, 3-HH), 3.37 (1H, quin.d, J 6.8, 2.3 Hz, 2-H); δC (126 MHz, CDCl3) 11.2 (CH3), 23.5 (CH2), 26.0 (CH2), 34.4 (CH2), 48.2 (CH2), 48.8 (CH2), 51.5 (CH2), 53.6 (CH), 57.2 (CH), 209.7 (C); m/z (ESI) 168 (MH+, 100%).(8R,10R)-8-Methyl-10-undecyl-1,5-dithia-9-azaspiro[5.5]undecane (13)
A solution of tert-butyl (2R,6R)-2-methyl-4-oxo-6-undecylpiperidine-1-carboxylate (11f) (0.0570 g, 0.136 mmol) in anhydrous dichloromethane (5 mL) was cooled to 0 °C and 1,3-propanedithiol (0.136 mL, 1.36 mmol) and boron trifluoride diethyl etherate (0.042 mL, 0.340 mmol) was added dropwise. The reaction was warmed to room temperature and stirred for 20 h. The reaction mixture was then diluted with dichloromethane (50 mL), washed with 1 M sodium hydroxide (50 mL) and brine (50 mL). The organic layer was dried (MgSO4) and concentrated in vacuo to give the crude product as a yellow oil. Purification by flash column chromatography using silica gel, eluting with 5% methanol in dichloromethane gave (8R,10R)-8-methyl-10-undecyl-1,5-dithia-9-azaspiro[5.5]undecane (13) as a colourless oil (0.0500 g, 90%). νmax/cm−1 (neat) 2922 (CH), 1465, 721; [α]24D +6.4 (c 1.0, CHCl3); δH (400 MHz, CDCl3) 0.88 (3H, t, J 6.9 Hz, 11′′-H3), 1.16–1.41 (21H, m, 1′-H3, 2′′-H2, 3′′-H2, 4′′-H2, 5′′-H2, 6′′-H2, 7′′-H2, 8′′-H2, 9′′-H2 and 10′′-H2), 1.58–1.76 (2H, m, 1′′-H2), 1.86 (1H, dd, J 14.0, 7.4 Hz, 7-HH), 1.90–2.04 (3H, m, 3-H2 and 11-HH), 2.24 (1H, dd, J 14.2, 4.4 Hz, 11-HH), 2.29 (1H, dd, J 14.0, 3.9 Hz, 7-HH), 2.77–2.94 (4H, m, 2-H2 and 4-H2), 3.08–3.21 (1H, m, 10-H), 3.27–3.41 (1H, m, 8-H); δC (101 MHz, CDCl3) 14.3 (CH3), 21.7 (CH3), 22.8 (CH2), 25.6 (CH2), 26.6 (2 × CH2), 26.8 (CH2), 29.7 (3 × CH2), 29.8 (2 × CH2), 32.1 (CH2), 35.1 (CH2), 41.3 (CH2), 44.0 (CH2), 45.0 (CH), 47.9 (CH2), 50.2 (CH); m/z (ESI) 358.2583 (MH+. C20H40NS2 requires 358.2597).(−)-Solenopsin A (3)14b
To a solution of (8R,10R)-8-methyl-10-undecyl-1,5-dithia-9-azaspiro[5.5]undecane (13) (0.0710 g, 0.155 mmol) in ethanol (10 mL) was added a suspension of RANEY® nickel (1.58 g) in ethanol (10 mL). The reaction mixture was heated under reflux whilst purging with hydrogen for 0.5 h. The reaction was then stirred under reflux, under an atmosphere of hydrogen for a further 3 h. After cooling to room temperature, the reaction mixture was filtered through a Celite pad and then concentrated in vacuo. The resulting residue was dissolved in dichloromethane (50 mL) and washed with 2 M sodium hydroxide (50 mL) and brine (50 mL). The organic layer was dried (MgSO4) and concentrated to give the crude product as a yellow oil. Purification by flash column chromatography using silica gel, eluting with 20% methanol in chloroform gave (−)-solenopsin A (3) as a colourless oil (0.0301 g, 77%). [α]25D −1.0 (c 0.5, MeOH), lit.14b [α]D −1.21 (c 0.94, MeOH); δH (400 MHz, CDCl3) 0.88 (3H, t, J 6.9 Hz, 11′′-H3), 1.07 (3H, d, J 6.5 Hz, 1′-H3), 1.19–1.70 (26H, m, 3-H2, 4-H2, 5-H2, 1′′-H2, 2′′-H2, 3′′-H2, 4′′-H2, 5′′-H2, 6′′-H2, 7′′-H2, 8′′-H2, 9′′-H2 and 10′′-H2), 2.87 (1H, qd, J 6.8, 4.4 Hz, 6-H), 3.06 (1H, quin.d, J 6.5, 3.6 Hz, 2-H); δC (101 MHz, CDCl3) 14.3 (CH3), 19.7 (CH2), 21.4 (CH3), 22.8 (CH2), 26.6 (CH2), 29.5 (CH2), 29.7 (3 × CH2), 29.8 (3 × CH2), 29.9 (CH2), 32.1 (CH2), 33.1 (CH2), 46.0 (CH), 51.0 (CH); m/z (EI) 253 (M+, 11%), 238 (36), 210 (5), 126 (6), 111 (8), 98 (100).Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from EPSRC (studentships to J. D. B., EP/N509176/1 and A. H. H., EP/K503058/1) and GlaxoSmithKline is gratefully acknowledged.Notes and references
- (a) Alkaloids: Chemical and Biological Perspectives, ed. S. W. Pelletier, Pergamon, Oxford, U.K., 1996, vol. 10 Search PubMed; (b) J. P. Michael, Nat. Prod. Rep., 2008, 25, 139 RSC.
- (a) P. Slosse and C. Hootelé, Tetrahedron Lett., 1978, 19, 397 CrossRef; (b) P. Slosse and C. Hootelé, Tetrahedron, 1981, 37, 4287 CrossRef.
- K. Fuji, T. Yamada, E. Fujita and H. Murata, Chem. Pharm. Bull., 1978, 26, 2515 CrossRef.
- (a) T. H. Jones, M. S. Blum and H. M. Fales, Tetrahedron, 1982, 38, 1949 CrossRef; (b) S. Leclercq, I. Thirionet, F. Broeders, D. Daloze, R. Vander Meer and J. C. Braekman, Tetrahedron, 1994, 50, 8465 CrossRef.
- J. L. Arbiser, T. Kau, M. Konar, K. Narra, R. Ramchandran, S. A. Summers, C. J. Vlahos, K. Ye, B. N. Perry, W. Matter, A. Fischl, J. Cook, P. A. Silver, J. Bain, P. Cohen, D. Whitmire, S. Furness, B. Govindarajan and J. P. Bowen, Blood, 2007, 109, 560 CrossRef PubMed.
- I. Karlsson, X. Zhou, R. Thomas, A. T. Smith, M. Y. Bonner, P. Bakshi, A. K. Banga, J. P. Bowen, G. Qabaja, S. L. Ford, M. D. Ballard, K. S. Peterson, X. G. Chen, B. Ogretmen, J. Zhang, E. B. Watkins, R. S. Arnold and J. L. Arbiser, Vasc. Cell, 2015, 7 DOI:10.1186/s13221-015-0030-2.
- (a) G. Ratle, X. Monseur, B. C. Das, J. Yassi, Q. Khuong-Huu and R. Goutarel, Bull. Soc. Chim. Fr., 1966, 2945 Search PubMed; (b) Q. Khuong-Huu, G. Ratle, X. Monseur and R. Goutarel, Bull. Soc. Chim. Belg., 1972, 81, 425 CrossRef.
- P. Bourrinet and A. Quevauviller, Ann. Pharm. Fr., 1968, 26, 787 Search PubMed.
- For recent reviews, see: (a) N. Veerasamy and R. G. Carter, Tetrahedron, 2016, 72, 4989 CrossRef; (b) N. Kandepedu, I. Abrunhosa-Thomas and Y. Troin, Org. Chem. Front., 2017, 4, 1655 RSC.
- For selected recent examples, see: (a) C. Gnamm, K. Brödner, C. M. Krauter and G. Helmchen, Chem. – Eur. J., 2009, 15, 10514 CrossRef PubMed; (b) V. H. Vu, F. Louafi, N. Girard, R. Marion, T. Roisnel, V. Dorcet and J.-P. Hurvois, J. Org. Chem., 2014, 79, 3358 CrossRef PubMed; (c) Y. Ying, H. Kim and J. Hong, Org. Lett., 2011, 13, 796 CrossRef PubMed; (d) H. Li and J. Wu, Org. Lett., 2015, 17, 5424 CrossRef PubMed; (e) T. Hirama, T. Umemura, N. Kogure, M. Kitajima and H. Takayama, Tetrahedron Lett., 2017, 58, 223 CrossRef; (f) W. Chen, L. Ma, A. Paul and D. Seidel, Nat. Chem., 2018, 10, 165 CrossRef PubMed.
- J. E. Baldwin, J. Chem. Soc., Chem. Commun., 1976, 734 RSC.
- (a) M. G. Banwell, B. D. Bissett, C. T. Bui, H. T. T. Pham and G. W. Simpson, Aust. J. Chem., 1998, 51, 9 CrossRef; (b) E. L. Wynne, G. J. Clarkson and M. Shipman, Tetrahedron Lett., 2008, 49, 250 CrossRef.
- L. S. Fowler, L. H. Thomas, D. Ellis and A. Sutherland, Chem. Commun., 2011, 47, 6569 RSC.
- (a) I. Abrunhosa-Thomas, O. Roy, M. Barra, T. Besset, P. Chalard and Y. Troin, Synlett, 2007, 1613 Search PubMed; (b) I. Abrunhosa-Thomas, A. Plas, A. Vogrig, N. Kandepedu, P. Chalard and Y. Troin, J. Org. Chem., 2013, 78, 2511 CrossRef PubMed.
- M. Daly, A. A. Cant, L. S. Fowler, G. L. Simpson, H. M. Senn and A. Sutherland, J. Org. Chem., 2012, 77, 10001 CrossRef PubMed.
- A. H. Harkiss and A. Sutherland, J. Org. Chem., 2018, 83, 535 CrossRef PubMed.
- C. S. Dexter, R. F. W. Jackson and J. Elliott, J. Org. Chem., 1999, 64, 7579 CrossRef.
- T. Hjelmgaard and D. Tanner, Org. Biomol. Chem., 2006, 4, 1796 RSC.
- The addition of Hünig's base during the hydrodehalogenation neutralises hydrogen iodide formed during this process, thus preventing poisoning of the Pd/C catalyst. See: (a) Y.-Y. Ku, Y.-M. Pu, T. Grieme, P. Sharma, A. V. Bhatia and M. Cowart, Tetrahedron, 2006, 62, 4584 CrossRef; (b) P. K. Mandal, J. S. Birtwistle and J. S. McMurray, J. Org. Chem., 2014, 79, 8422 CrossRef PubMed.
- L. S. Fowler, D. Ellis and A. Sutherland, Org. Biomol. Chem., 2009, 7, 4309 RSC.
- A similar cyclisation that initially produces the kinetic 2,6-trans isomer and then the thermodynamic 2,6-cis product over an extended time period, via a retro-conjugate addition process was also observed by Troin and co-workers in their intramolecular Michael-type reaction of carbamate-protected amine-substituted enones. See ref. 14b.
- It is well established that 2,6-cis-piperidines are the more stable, thermodynamic isomer, while 2,6-trans-piperidines are generated under kinetic conditions. See ref. 14b and: (a) S. Ciblat, P. Calinaud, J.-L. Canet and Y. Troin, J. Chem. Soc., Perkin Trans. 1, 2000, 353 RSC; (b) J. Quick, C. Mondello, M. Humora and T. Brennan, J. Org. Chem., 1978, 43, 2705 CrossRef.
- T. Markidis and G. Kokotos, J. Org. Chem., 2001, 66, 1919 CrossRef PubMed.
- K. B. Hansen, T. Rosner, M. Kubryk, P. G. Dormer and J. D. Armstrong, Org. Lett., 2005, 7, 4935 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra for all compounds. See DOI: 10.1039/c8ob01363b |
| This journal is © The Royal Society of Chemistry 2018 |